
A Molecular Phylogeny of Stylodipus (Dipodidae, Mammalia): A Small Genus with a Complex History

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Sampling, DNA Extraction, Amplification, and Sequencing
2.2. Phylogenetic Analysis
2.3. Molecular Dating and Species Tree Reconstruction
2.4. Biogeographic Paleo-Reconstructions
3. Results
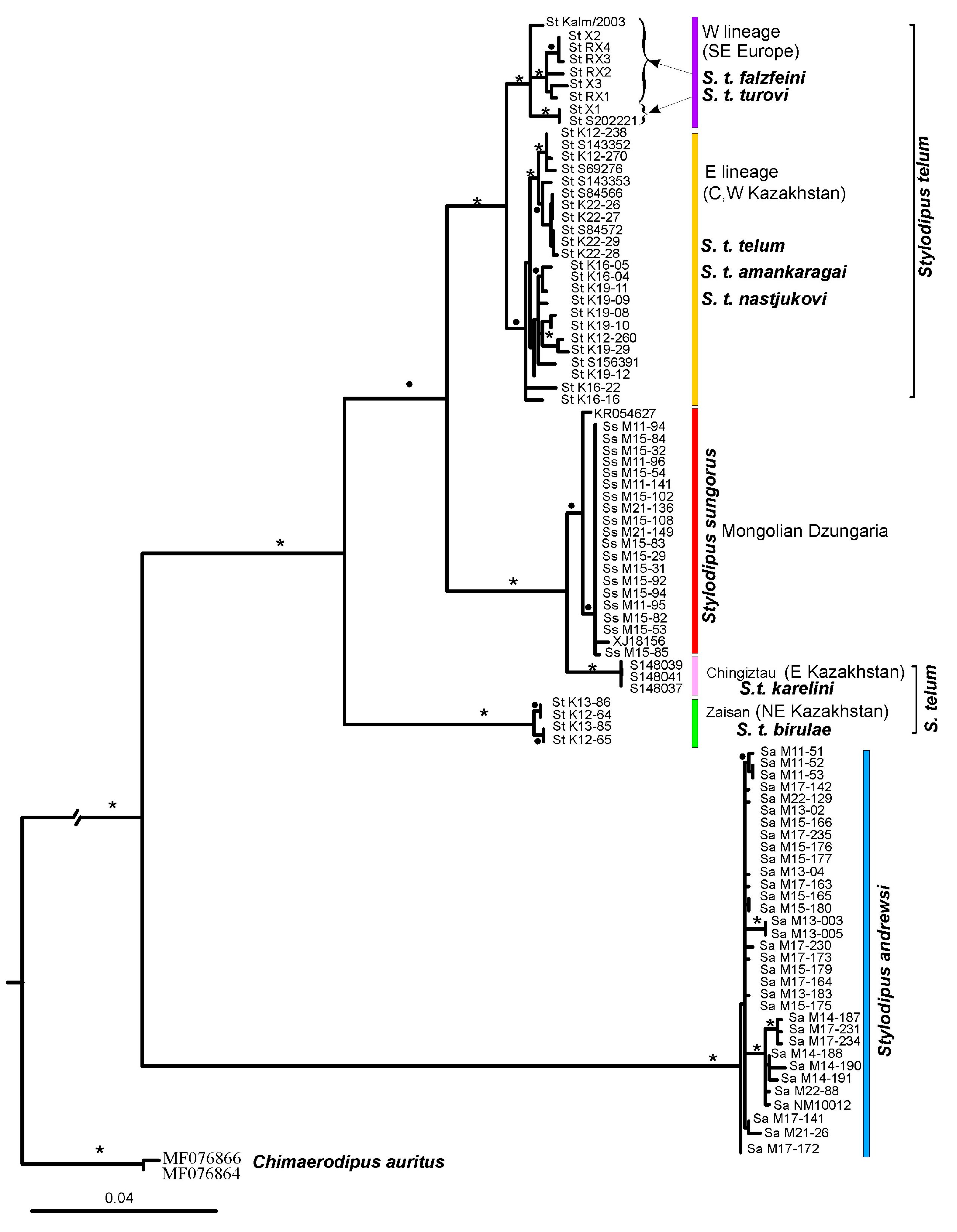
3.1. Cytb Tree and Mitochondrial Variability
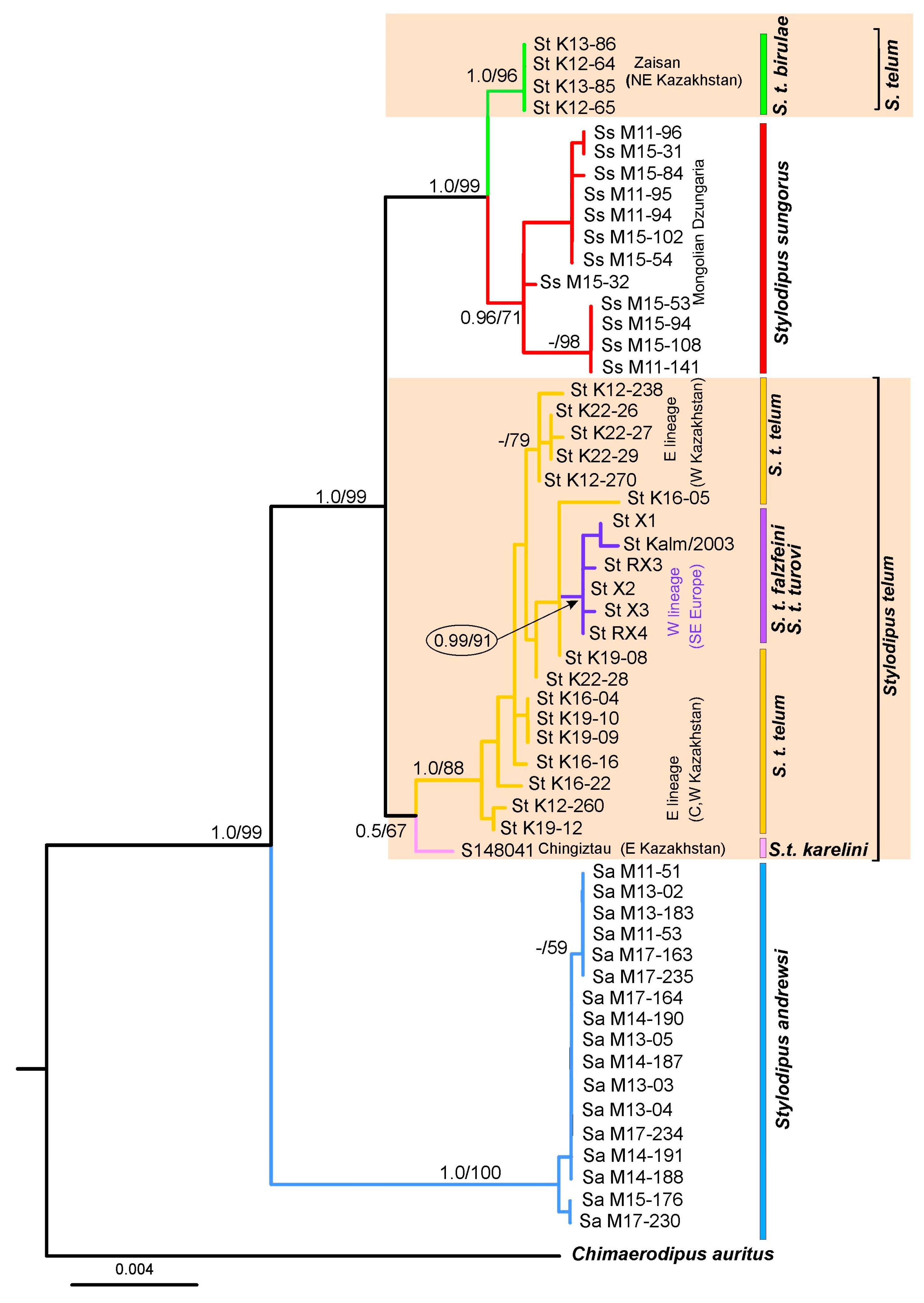
3.2. Nuclear Trees and Networks
3.3. Analysis of Nuclear Concatenation
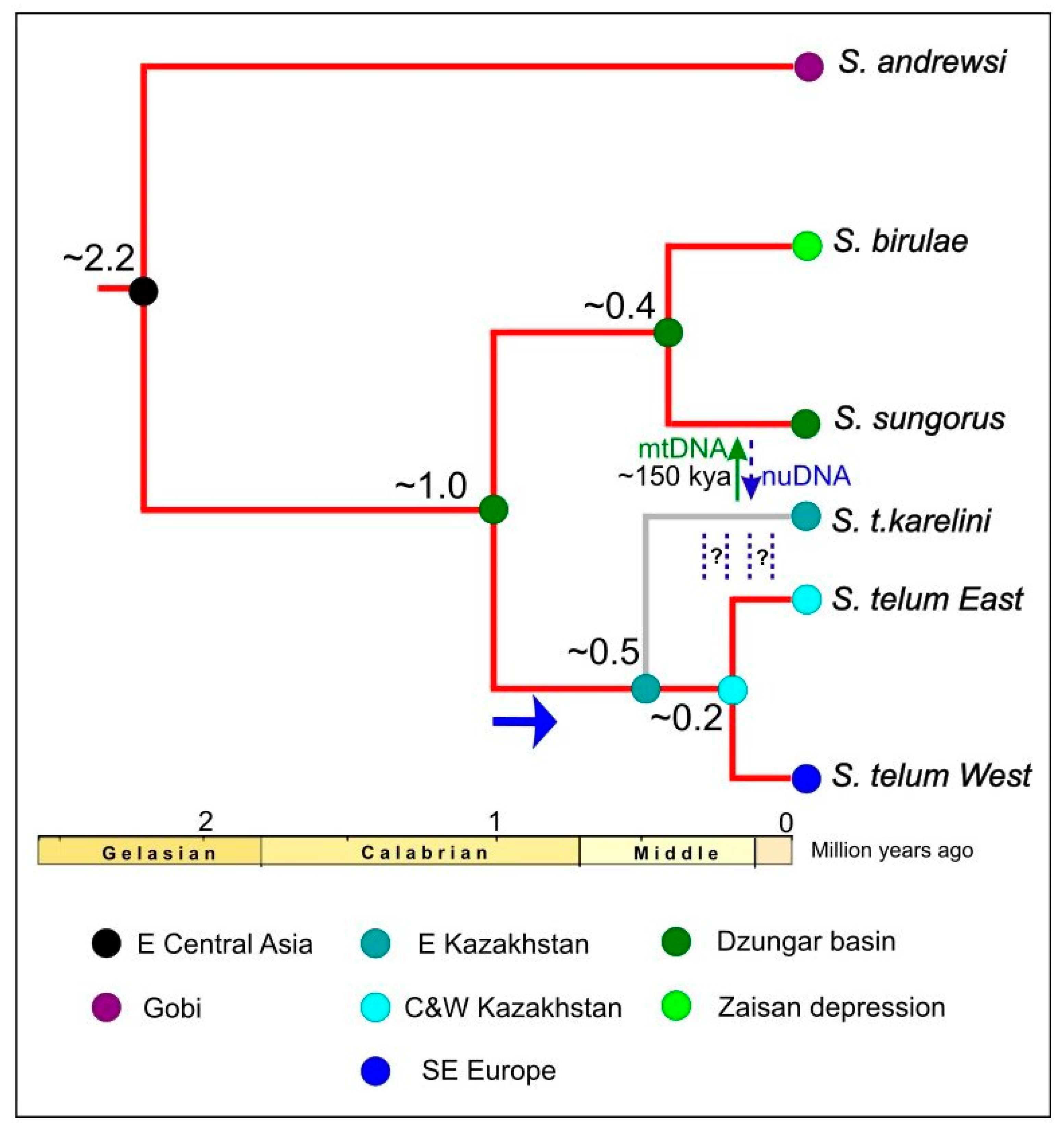
3.4. Species Tree and Divergence Times
3.5. Biogeographic Paleo-Reconstructions
4. Discussion
4.1. Genetic Diversity in Genus Stylodipus and Implications for Its Taxonomy
4.2. An Evolutionary Biogeographic Scenario for Stylodipus
4.3. Intraspecific Variation and Phylogeographic Patterns
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shenbrot, G.I.; Sokolov, V.E.; Heptner, V.G.; Kovalskaya, Y.M. Jerboas. Mammals of Russia and Adjacent Regions; Hoffmann, R.S., Wilson, D.E., Eds.; Science Publishers, Enfield: Enfield, NH, USA, 2008. [Google Scholar]
- Sokolov, V.E.; Lobachev, V.S.; Orlov, V.N. Mammals of Mongolia. Jerboas: Dipodinae, Allactaginae; Nauka Press: Moscow, Russia, 1998. (In Russian) [Google Scholar]
- Michaux, J.; Shenbrot, G. Family Dipodidae (Jerboas). In Handbook of the Mammals of the World, 7. Rodents II; Wilson, D.E., Lacher, T.E., Mittermeier, R.A., Eds.; Lynx Edicions in association with Conservation International and IUCN: Barselona, Spain, 2017; pp. 62–100. [Google Scholar]
- Shenbrot, G.I. Subspecific Taxonomy Revision of Common Thick-Tailed Three-Toed Jerboa, Stylodipus Telum (Rodentia, Dipodidae). Zool. Z. 1991, 70, 118–127. [Google Scholar]
- Shenbrot, G.; Bannikova, A.; Giraudoux, P.; Quéré, J.-P.; Raoul, F.; Lebedev, V. A New Recent Genus and Species of Three-Toed Jerboas (Rodentia: Dipodinae) from China: A Living Fossil? J. Zool. Syst. Evol. Res. 2017, 55, 356–368. [Google Scholar] [CrossRef]
- Pisano, J.; Condamine, F.L.; Lebedev, V.; Bannikova, A.; Quéré, J.-P.; Shenbrot, G.I.; Pagès, M.; Michaux, J.R. Out of Himalaya: The Impact of Past Asian Environmental Changes on the Evolutionary and Biogeographical History of Dipodoidea (Rodentia). J. Biogeogr. 2015, 42, 856–870. [Google Scholar] [CrossRef]
- Rusin, M. Phylogeography of the Western Populations of Stylodipus Telum (Rodentia, Dipodidae) Based on Mitochondrial DNA. Zoodiversity 2023, 57, 13–18. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor: New York, NY, USA, 1989. [Google Scholar]
- Montgelard, C.; Bentz, S.; Tirard, C.; Verneau, O.; Catzeflis, F.M. Molecular Systematics of Sciurognathi (Rodentia): The Mitochondrial Cytochrome b and 12S RRNA Genes Support the Anomaluroidea (Pedetidae and Anomaluridae). Mol. Phylogenetics Evol. 2002, 22, 220–233. [Google Scholar] [CrossRef]
- Lebedev, V.S.; Bannikova, A.A.; Lu, L.; Snytnikov, E.A.; Adiya, Y.; Solovyeva, E.N.; Abramov, A.V.; Surov, A.V.; Shenbrot, G.I. Phylogeographical Study Reveals High Genetic Diversity in a Widespread Desert Rodent, Dipus Sagitta (Dipodidae: Rodentia). Biol. J. Linn. Soc. 2018, 123, 445–462. [Google Scholar] [CrossRef]
- Lebedev, V.S.; Bannikova, A.A.; Pagès, M.; Pisano, J.; Michaux, J.R.; Shenbrot, G.I. Molecular Phylogeny and Systematics of Dipodoidea: A Test of Morphology-Based Hypotheses. Zool. Scr. 2013, 42, 231–249. [Google Scholar] [CrossRef]
- Lebedev, V.S.; Shenbrot, G.I.; Krystufek, B.; Mahmoudi, A.; Melnikova, M.N.; Solovyeva, E.N.; Lisenkova, A.A.; Undrakhbayar, E.; Rogovin, K.A.; Surov, A.V.; et al. Phylogenetic Relations and Range History of Jerboas of the Allactaginae Subfamily (Dipodidae, Rodentia). Sci. Rep. 2022, 12, 842. [Google Scholar] [CrossRef]
- Yang, D.Y.; Eng, B.; Waye, J.S.; Dudar, J.C.; Saunders, S.R. Improved DNA Extraction from Ancient Bones Using Silica-Based Spin Columns. Am. J. Phys. Anthropol. 1998, 105, 539–543. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Stephens, M.; Donnelly, P. A Comparison of Bayesian Methods for Haplotype Reconstruction from Population Genotype Data. Am. J. Hum. Genet. 2003, 73, 1162–1169. [Google Scholar] [CrossRef] [PubMed]
- Stephens, M.; Smith, N.J.; Donnelly, P. A New Statistical Method for Haplotype Reconstruction from Population Data. Am. J. Hum. Genet. 2001, 68, 978–989. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A Software for Comprehensive Analysis of DNA Polymorphism Data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. POPART: Full-feature Software for Haplotype Network Construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Douglas, J.; Jiménez-Silva, C.L.; Bouckaert, R. StarBeast3: Adaptive Parallelized Bayesian Inference under the Multispecies Coalescent. Syst. Biol. 2022, 71, 901–916. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, H.A.; Heled, J.; Xie, D.; Drummond, A.J. Computational Performance and Statistical Accuracy of *BEAST and Comparisons with Other Methods. Syst. Biol. 2016, 65, 381–396. [Google Scholar] [CrossRef]
- Peterson, A.T.; Soberón, J.; Sánchez-Cordero, V. Conservatism of Ecological Niches in Evolutionary Time. Science 1999, 285, 1265–1267. [Google Scholar] [CrossRef]
- Fick, S.E.; Hijmans, R.J. WorldClim 2: New 1-km Spatial Resolution Climate Surfaces for Global Land Areas. Int. J. Climatol. 2017, 37, 4302–4315. [Google Scholar] [CrossRef]
- Gamisch, A. Oscillayers: A Dataset for the Study of Climatic Oscillations over Plio-Pleistocene Time-scales at High Spatial-temporal Resolution. Glob. Ecol. Biogeogr. 2019, 28, 1552–1560. [Google Scholar] [CrossRef]
- Anderson, R.P.; Raza, A. The Effect of the Extent of the Study Region on GIS Models of Species Geographic Distributions and Estimates of Niche Evolution: Preliminary Tests with Montane Rodents (Genus Nephelomys) in Venezuela. J. Biogeogr. 2010, 37, 1378–1393. [Google Scholar] [CrossRef]
- Elith, J.; Phillips, S.J.; Hastie, T.; Dudík, M.; Chee, Y.E.; Yates, C.J. A Statistical Explanation of MaxEnt for Ecologists. Divers. Distrib. 2011, 17, 43–57. [Google Scholar] [CrossRef]
- Phillips, S.J.; Anderson, R.P.; Schapire, R.E. Maximum Entropy Modeling of Species Geographic Distributions. Ecol. Modell. 2006, 190, 231–259. [Google Scholar] [CrossRef]
- Phillips, S.J.; Dudík, M. Modeling of Species Distributions with Maxent: New Extensions and a Comprehensive Evaluation. Ecography 2008, 31, 161–175. [Google Scholar] [CrossRef]
- Liu, C.; White, M.; Newell, G. Selecting Thresholds for the Prediction of Species Occurrence with Presence-Only Data. J. Biogeogr. 2013, 40, 778–789. [Google Scholar] [CrossRef]
- Yu, Y.; Harris, A.J.; Blair, C.; He, X. RASP (Reconstruct Ancestral State in Phylogenies): A tool for historical biogeography. Mol. Phylogenetics Evol. 2015, 87, 46–49. [Google Scholar] [CrossRef]
- Molak, M.; Ho, S.Y.W. Prolonged Decay of Molecular Rate Estimates for Metazoan Mitochondrial DNA. PeerJ 2015, 3, e821. [Google Scholar] [CrossRef] [PubMed]
- Lisenkova, A.A.; Lebedev, V.S.; Undrakhbayar, E.; Bogatyreva, V.Y.; Melnikova, M.N.; Nazarov, R.A.; Rogovin, K.A.; Surov, A.V.; Shenbrot, G.I.; Bannikova, A.A. Phylogeny of the Dipus Sagitta Species Complex by Nuclear Gene Sequences. Dokl. Biol. Sci. 2023, 509, 135–139. [Google Scholar] [CrossRef]
- Bannikova, A.; Lebedev, V.; Dubrovskaya, A.; Solovyeva, E.; Moskalenko, V.; Kryštufek, B.; Hutterer, R.; Bykova, E.; Zhumabekova, B.; Rogovin, K.; et al. Genetic Evidence for Several Cryptic Species within the Scarturus Elater Species Complex (Rodentia: Dipodoidea): When Cryptic Species Are Really Cryptic. Biol. J. Linn. Soc. 2019, 126, 16–39. [Google Scholar] [CrossRef]
- Nanova, O.G.; Lebedev, V.S.; Matrosova, V.A.; Adiya, Y.; Undrakhbayar, E.; Surov, A.V.; Shenbrot, G.I. Phylogeography, Phylogeny, and Taxonomical Revision of the Midday Jird (Meriones meridianus) Species Complex from Dzungaria. J. Zool. Syst. Evol. Res. 2020, 58, 1335–1358. [Google Scholar] [CrossRef]
- Sokolov, V.E.; Shebrot, G.I. A New Species, Mongolian Three-Toed Jerboa Stylodipus Sungorus Sp. n. (Rodentia, Dipodidae) from Western Mongolia. Zool. Zhurnal 1987, 66, 579–587. [Google Scholar]
- Lebedev, V.; Bogdanov, A.; Brandler, O.; Melnikova, M.; Enkhbat, U.; Tukhbatullin, A.; Abramov, A.; Surov, A.; Bakloushinskaya, I.; Bannikova, A. Cryptic variation in mole voles Ellobius (Arvicolinae, Rodentia) of Mongolia. Zool. Scr. 2020, 49, 535–548. [Google Scholar] [CrossRef]
- Balmori-de la Puente, A.; Ventura, J.; Miñarro, M.; Somoano, A.; Hey, J.; Castresana, J. Divergence time estimation using ddRAD data and an isolation-with-migration model applied to water vole populations of Arvicola. Sci. Rep. 2022, 12, 4065. [Google Scholar] [CrossRef]
- Romanenko, S.A.; Lebedev, V.S.; Serdukova, N.A.; Feoktistova, N.Y.; Surov, A.V.; Graphodatsky, A.S. Comparative Cytogenetics of Hamsters of the Genus Allocricetulus Argyropulo 1932 (Cricetidae, Rodentia). Cytogenet. Genome Res. 2013, 139, 258–266. [Google Scholar] [CrossRef]
- Bannikova, A.A.; Lebedev, V.S.; Poplavskaya, N.S.; Simanovsky, S.A.; Undrakhbayar, E.; Adiya, Y.; Surov, A.S. Phylogeny and Phylogeography of Arvicoline and Lagurine Voles of Mongolia. Folia Zool. 2019, 68, 100–113. [Google Scholar] [CrossRef]
- Zazhigin, V.S.; Lopatin, A.V. The History of the Dipodoidea (Rodentia, Mammalia) in the Miocene of Asia: 4. Dipodinae at the Miocene-Pliocene Transition. Paleontol. J. 2001, 35, 60–74. [Google Scholar]
- Shenbrot, G.I.; Krasnov, B.R.; Rogovin, K.A. Spatial Ecology of Desert Rodent Communities; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 1999. [Google Scholar]
- Kapustina, S.; Adiya, Y.; Lyapunova, E.; Brandler, O. Phylogeography of the Pallid Ground Squirrel (Spermophilus Pallidicauda; Satunin, 1903) as a Consequence of Holocene Changes in the Mongolian Steppe Ecosystems. bioRxiv 2022, 2022-11. [Google Scholar] [CrossRef]
- Cheng, J.; Xia, L.; Feijó, A.; Shenbrot, G.I.; Wen, Z.; Ge, D.; Lu, L.; Yang, Q. Phylogeny, Taxonomic Reassessment and ‘Ecomorph’ Relationship of the Orientallactaga Sibirica Complex (Rodentia: Dipodidae: Allactaginae). Zool. J. Linn. Soc. 2021, 192, 185–205. [Google Scholar] [CrossRef]
- Huchon, D.; Madsen, O.; Sibbald, M.J.J.B.; Ament, K.; Stanhope, M.J.; Catzeflis, F.; de Jong, W.W.; Douzery, E.J.P. Rodent Phylogeny and a Timescale for the Evolution of Glires: Evidence from an Extensive Taxon Sampling Using Three Nuclear Genes. Mol. Biol. Evol. 2002, 19, 1053–1065. [Google Scholar] [CrossRef]
- Luo, G.; Ding, L.; Liao, J. The Complete Mitochondrial Genome of Stylodipus Telum (Rodentia: Dipodidae) and Its Phylogenetic Analysis. Mitochondrial DNA Part A 2016, 27, 2568–2569. [Google Scholar] [CrossRef]
- Wu, S.; Wu, W.; Zhang, F.; Ye, J.; Ni, X.; Sun, J.; Edwards, S.V.; Meng, J.; Organ, C.L. Molecular and Paleontological Evidence for a Post-Cretaceous Origin of Rodents. PLoS ONE 2012, 7, e46445. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Xia, L.; Kimura, Y.; Shenbrot, G.; Zhang, Z.; Ge, D.; Yang, Q. Tracing the Origin and Diversification of Dipodoidea (Order: Rodentia): Evidence from Fossil Record and Molecular Phylogeny. Evol. Biol. 2013, 40, 32–44. [Google Scholar] [CrossRef]
- Rusin, M.; Lebedev, V.S.; Matrosova, V.; Zemlemerova, E.D.; Lopatina, N.; Bannikova, A.A. Hidden Diversity in the Caucasian Mountains: An Example of Birch Mice (Rodentia, Sminthidae, Sicista). Hystrix Ital. J. Mammal. 2018, 29, 61–66. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Split | Age, MYA (Posterior Mean) | 95% HPD, MYA (Highest Posterior Distribution) |
|---|---|---|
| S. andrewsi/S. telum species complex | 2.23 | 1.64–2.8 |
| S. telum/S. sungorus + S. birulae | 1.04 | 0.66–1.43 |
| S. sungorus/S. birulae | 0.42 | 0.20–0.66 |
| S. telum East/S. telum West | 0.20 | 0.09–0.32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lebedev, V.S.; Mirzoyan, D.A.; Shenbrot, G.I.; Solovyeva, E.N.; Bogatyreva, V.Y.; Lisenkova, A.A.; Undrakhbayar, E.; Sukhchuluun, G.; Rogovin, K.A.; Surov, A.V.; et al. A Molecular Phylogeny of Stylodipus (Dipodidae, Mammalia): A Small Genus with a Complex History. Diversity 2023, 15, 1114. https://doi.org/10.3390/d15111114
Lebedev VS, Mirzoyan DA, Shenbrot GI, Solovyeva EN, Bogatyreva VY, Lisenkova AA, Undrakhbayar E, Sukhchuluun G, Rogovin KA, Surov AV, et al. A Molecular Phylogeny of Stylodipus (Dipodidae, Mammalia): A Small Genus with a Complex History. Diversity. 2023; 15(11):1114. https://doi.org/10.3390/d15111114
Chicago/Turabian StyleLebedev, Vladimir S., Daniil A. Mirzoyan, Georgy I. Shenbrot, Evgeniya N. Solovyeva, Varvara Yu. Bogatyreva, Alexandra A. Lisenkova, Enkhbat Undrakhbayar, Gansukh Sukhchuluun, Konstantin A. Rogovin, Alexei V. Surov, and et al. 2023. "A Molecular Phylogeny of Stylodipus (Dipodidae, Mammalia): A Small Genus with a Complex History" Diversity 15, no. 11: 1114. https://doi.org/10.3390/d15111114
APA StyleLebedev, V. S., Mirzoyan, D. A., Shenbrot, G. I., Solovyeva, E. N., Bogatyreva, V. Y., Lisenkova, A. A., Undrakhbayar, E., Sukhchuluun, G., Rogovin, K. A., Surov, A. V., & Bannikova, A. A. (2023). A Molecular Phylogeny of Stylodipus (Dipodidae, Mammalia): A Small Genus with a Complex History. Diversity, 15(11), 1114. https://doi.org/10.3390/d15111114