
The Diverse Reticulate Genetic Set-Up of Endangered Gladiolus palustris in Southern Germany Has Consequences for the Development of Conservation Strategies

Abstract
:1. Introduction
2. Materials and Methods
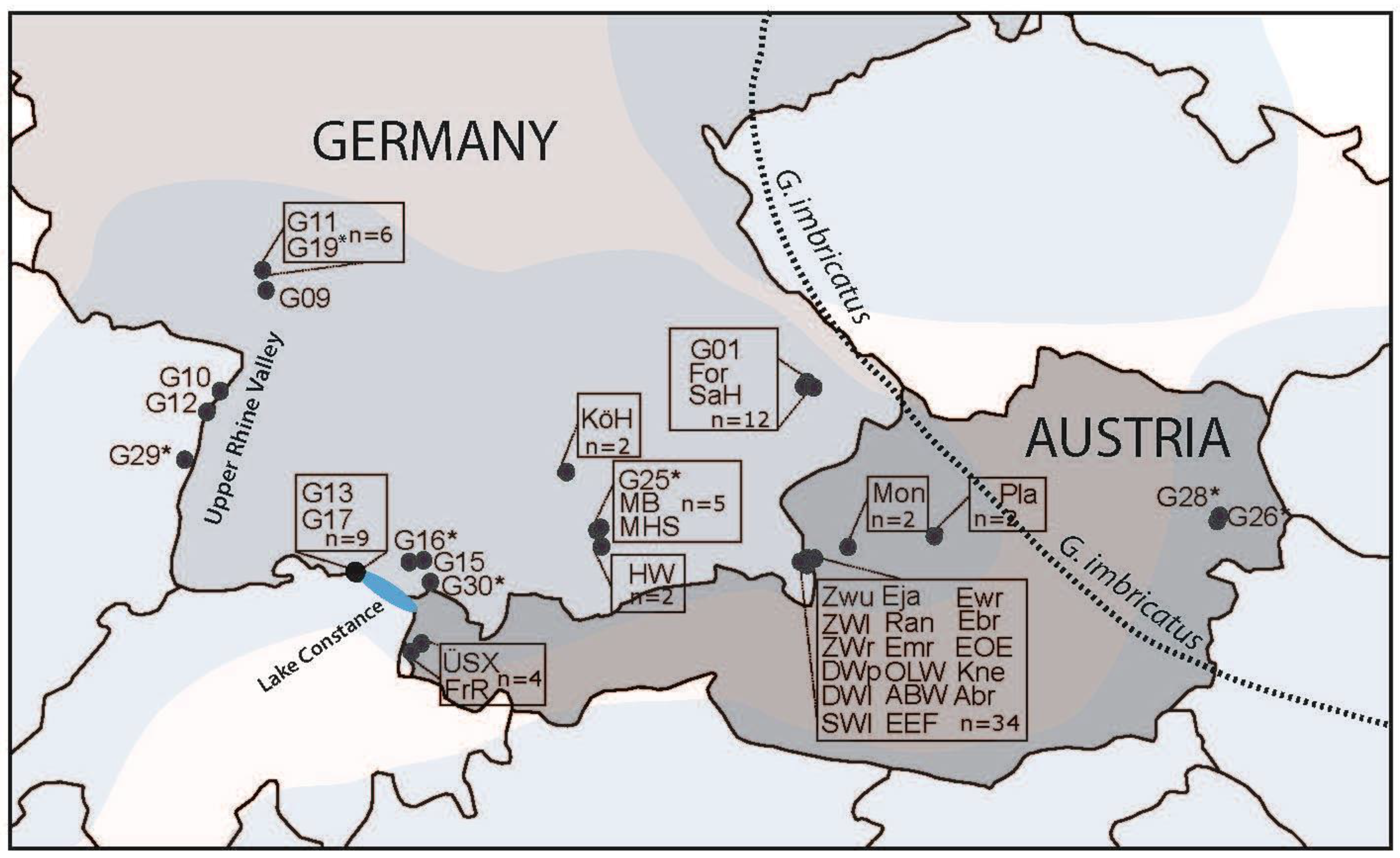
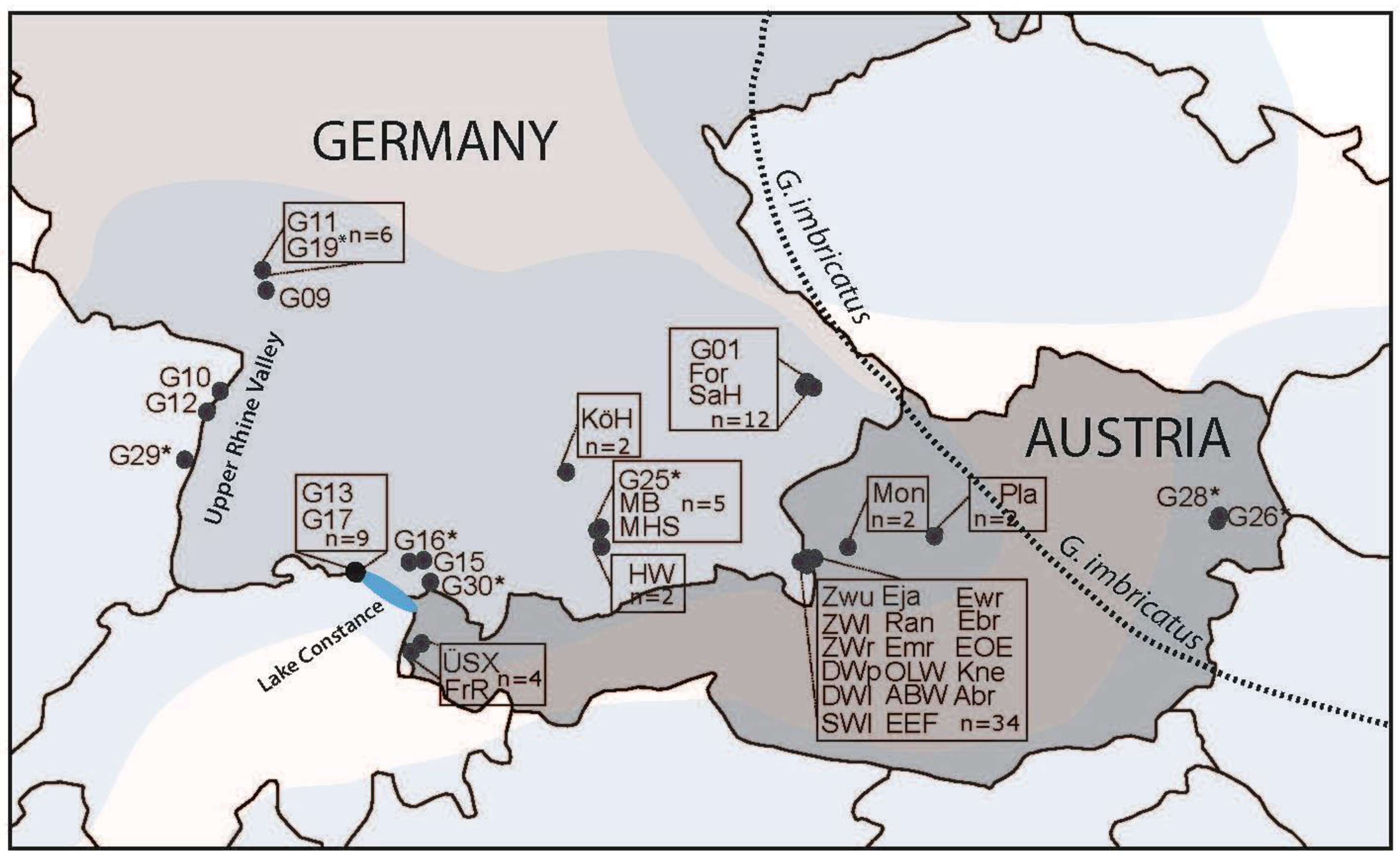
2.1. Focal Species, Study Area, Sampling, and Experimental Design
2.2. AFLP-Based Genotyping and Genetic Data Analyses
2.3. Species Assignment Using DNA Sequence Information from Internal Transcribed Spacer 1 and 2 (ITS) and Phylogenetic Analyses
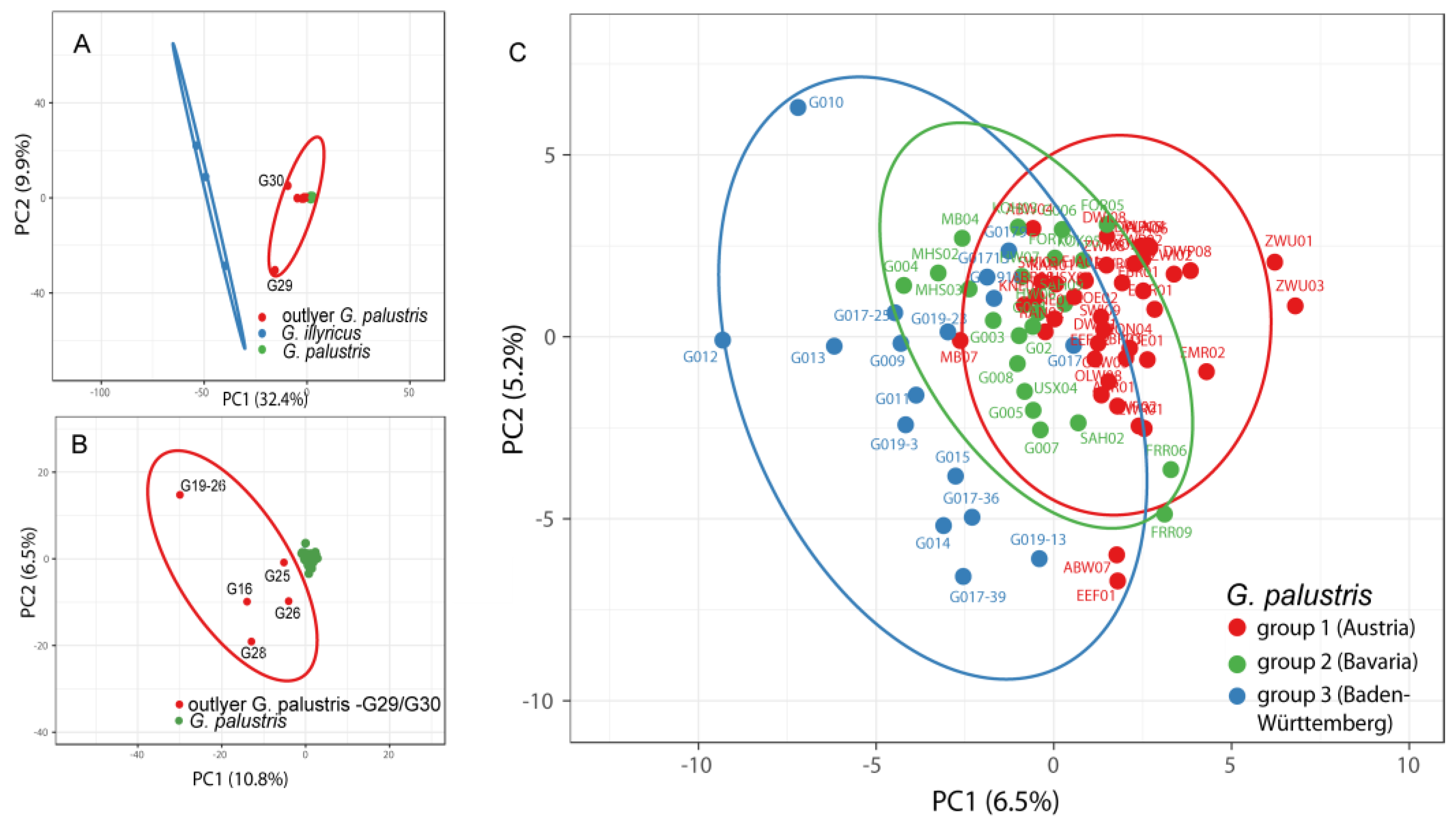
3. Results
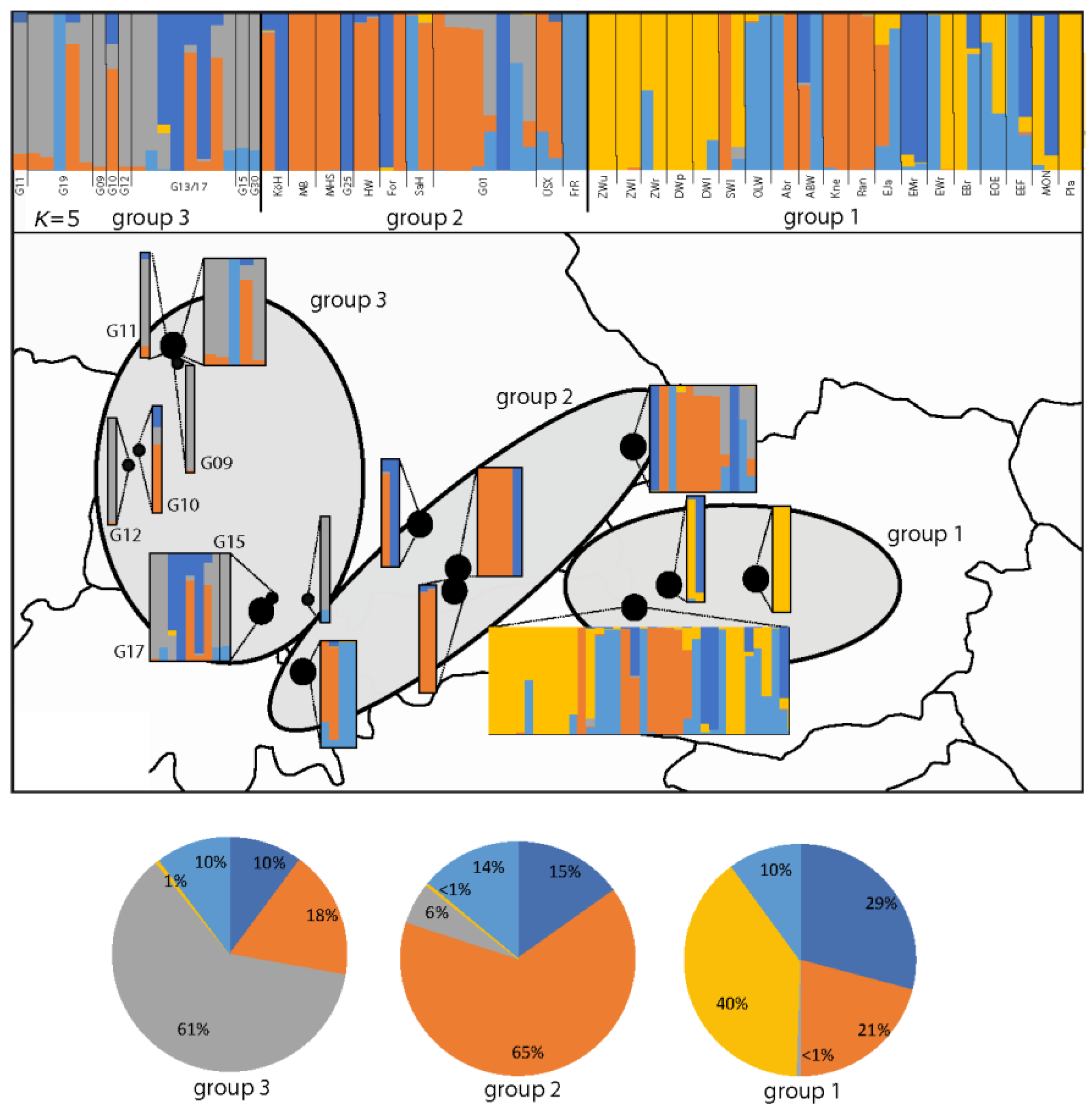
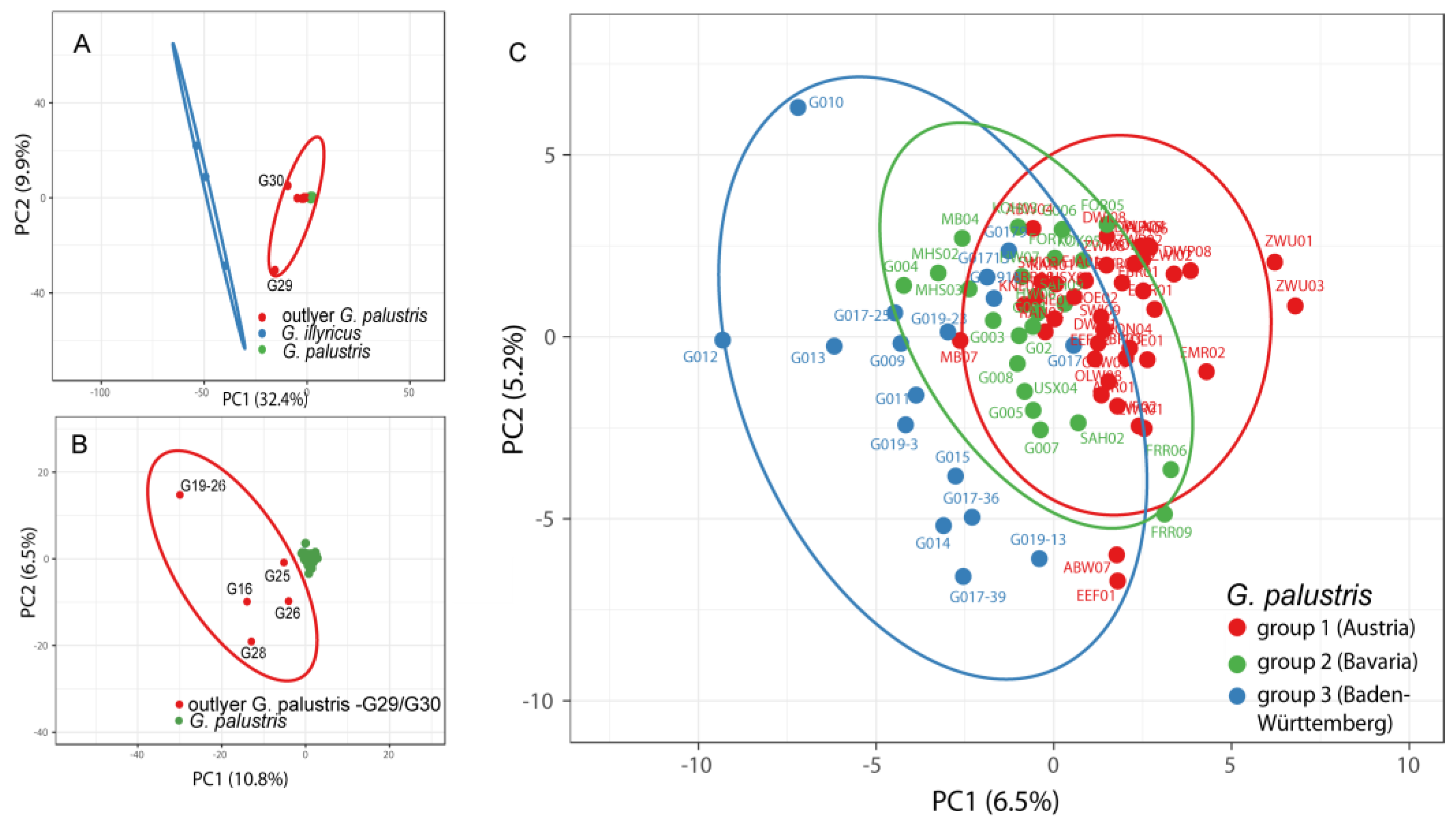
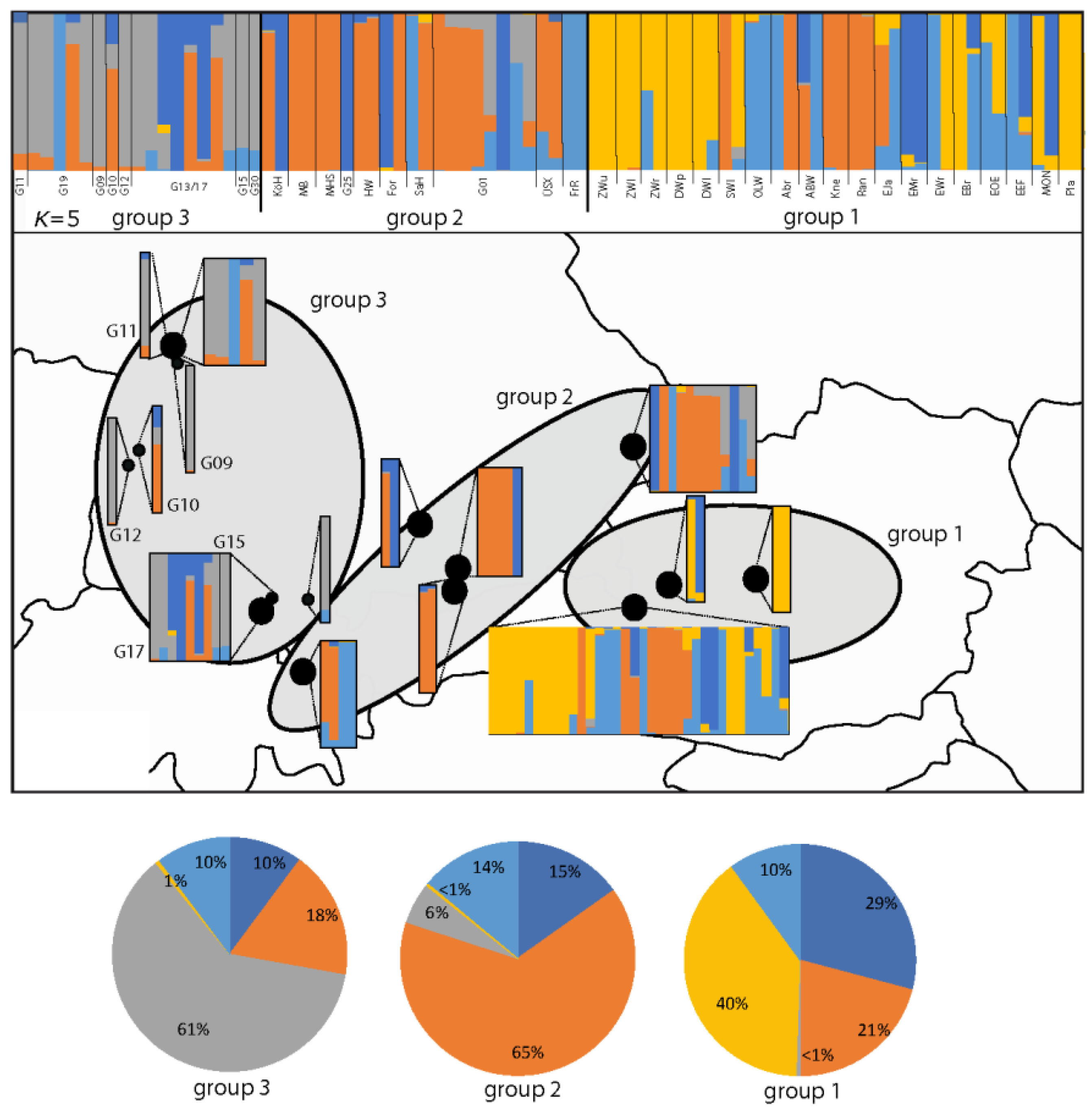
3.1. AFLP Analysis of Gladiolus Palustris Accessions
3.2. Genetic Variation within and among Regions of Gladiolus palustris Accessions
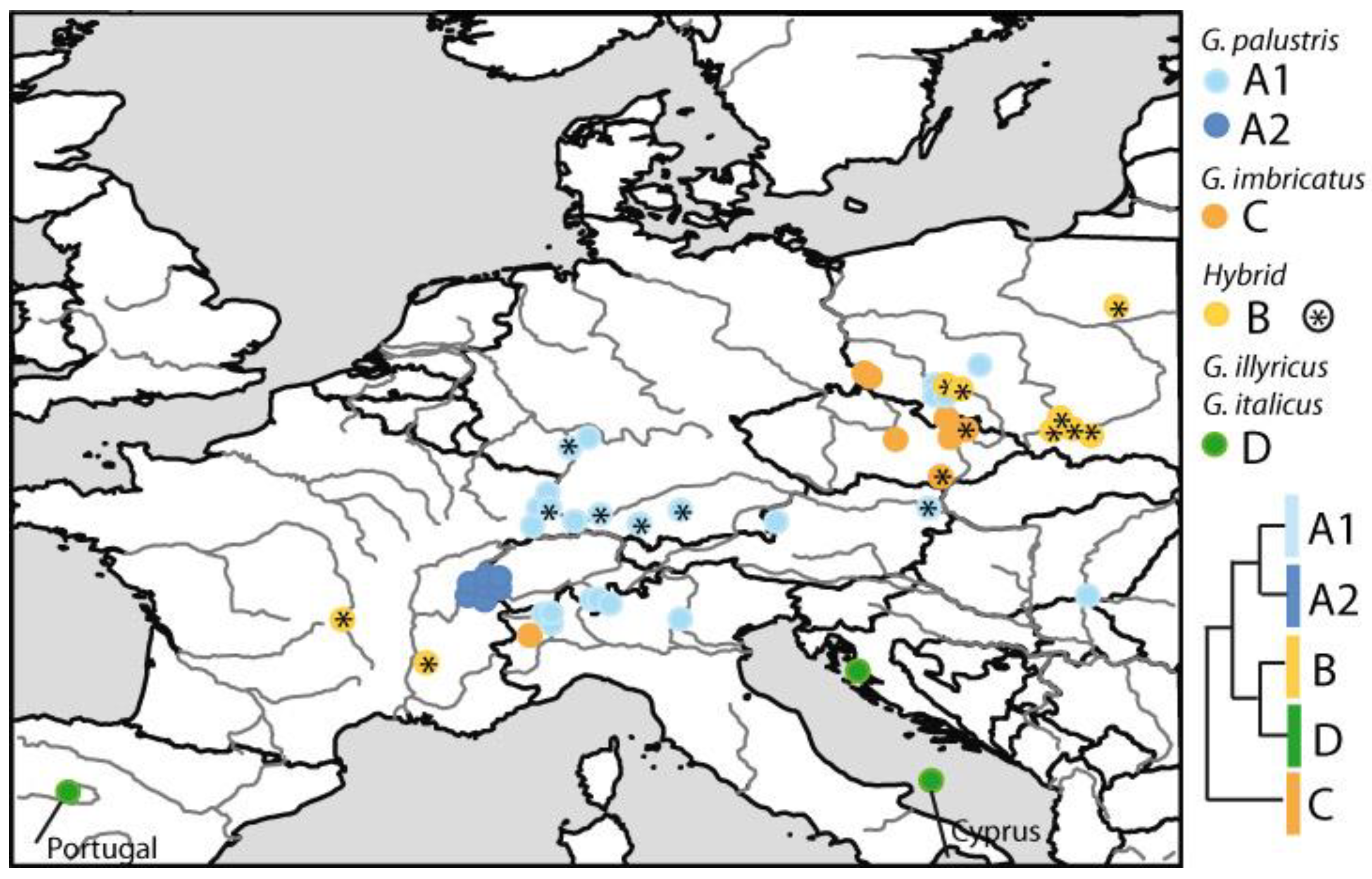
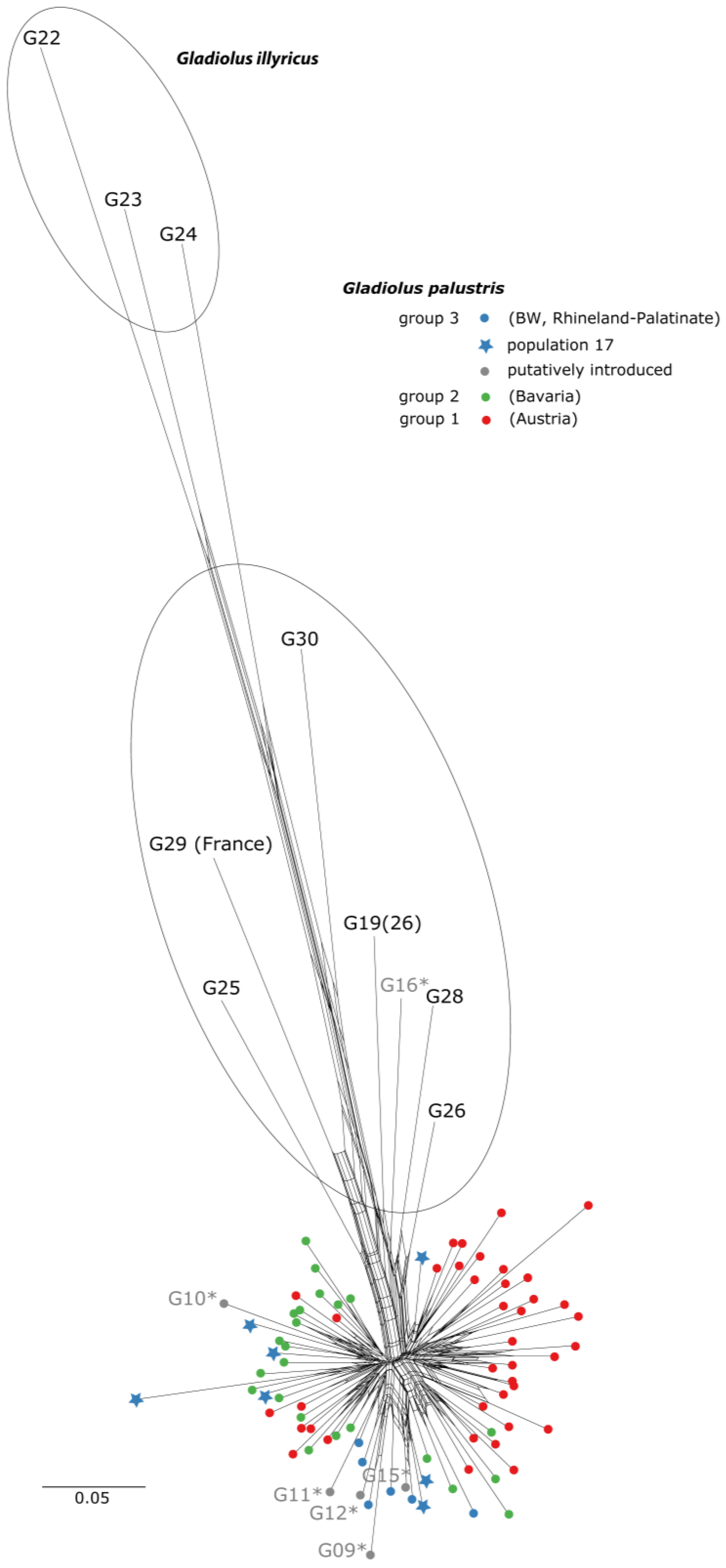
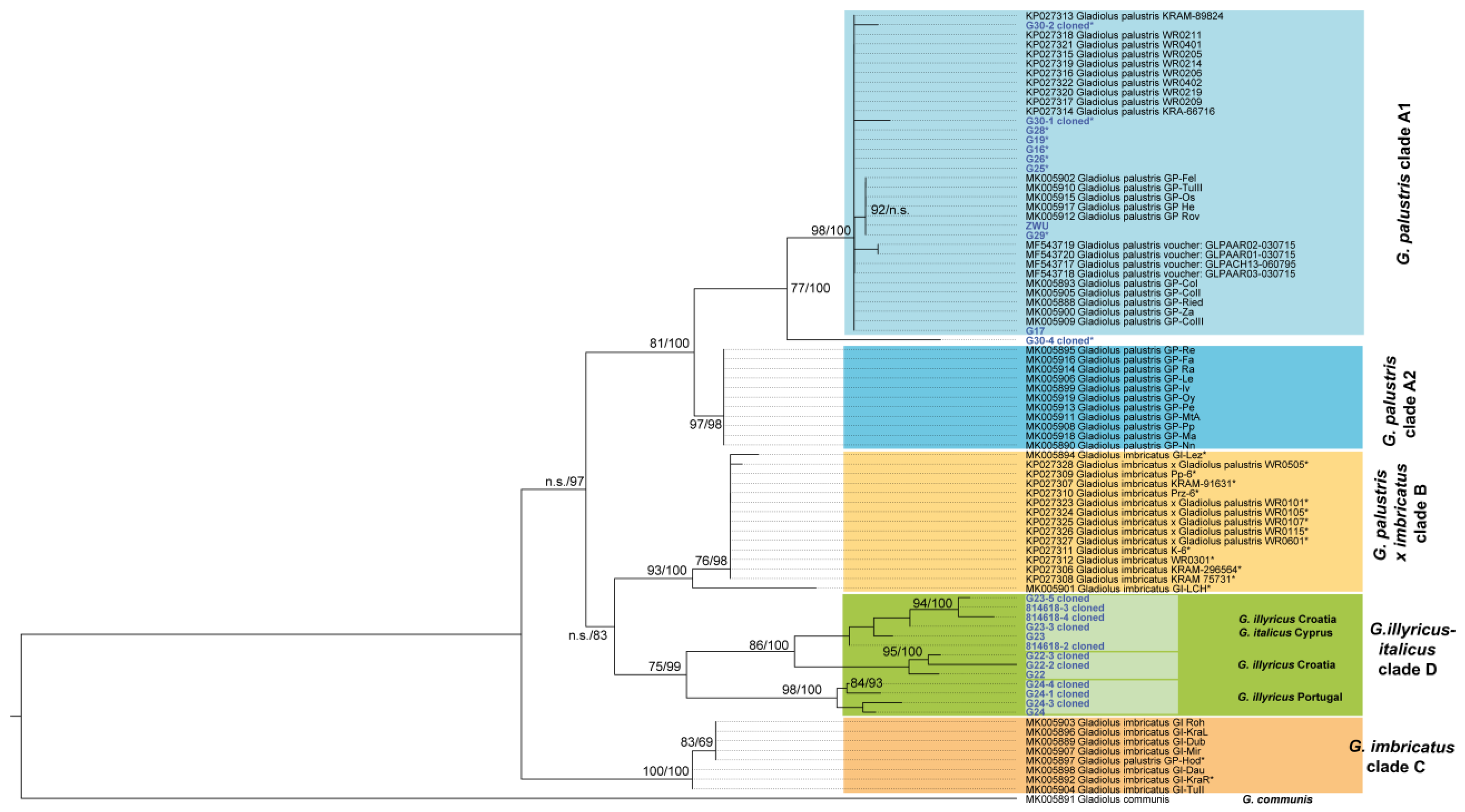
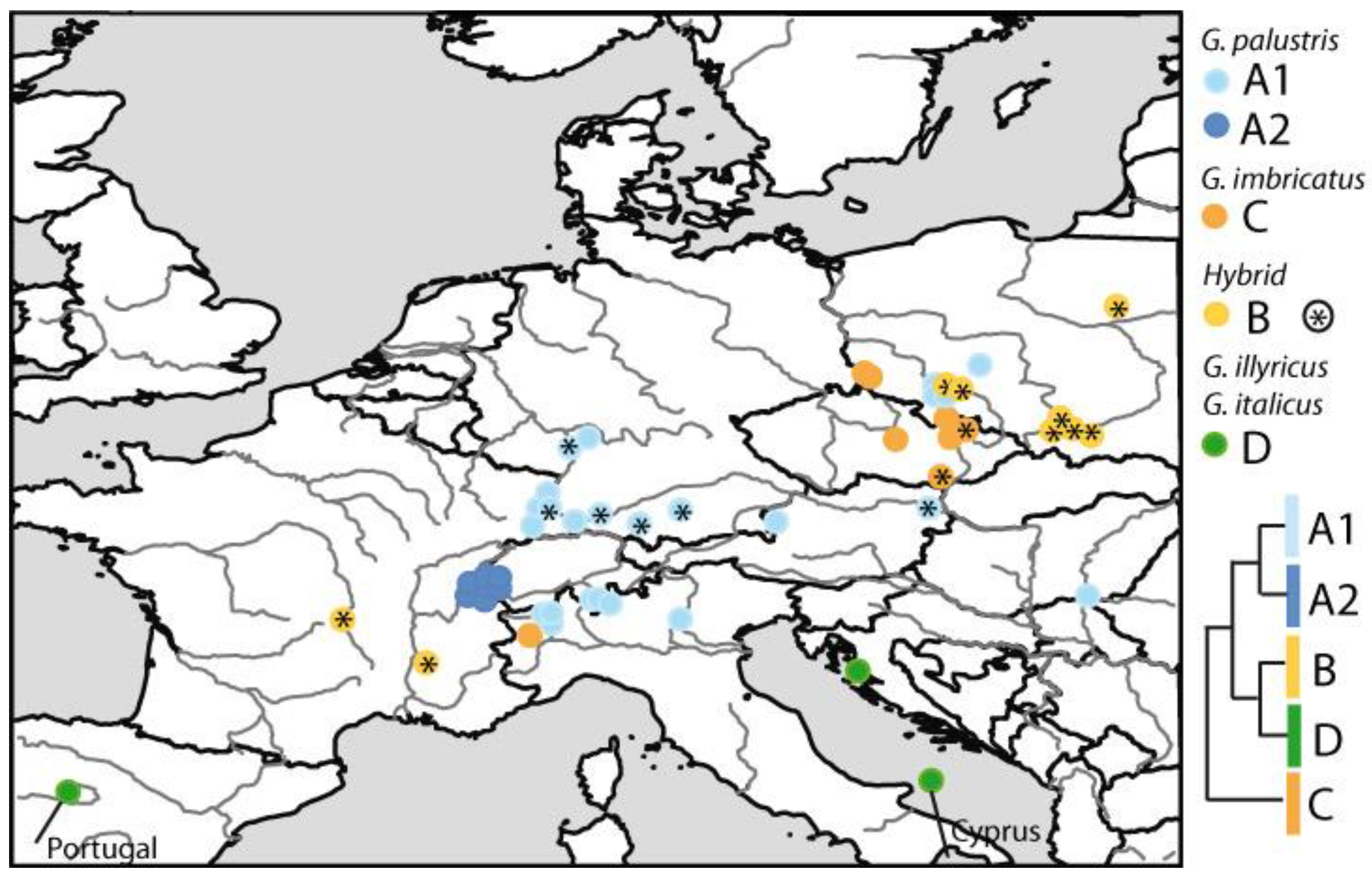
3.3. Phylogenetic Inference, Taxon Identity and Geographic Distribution
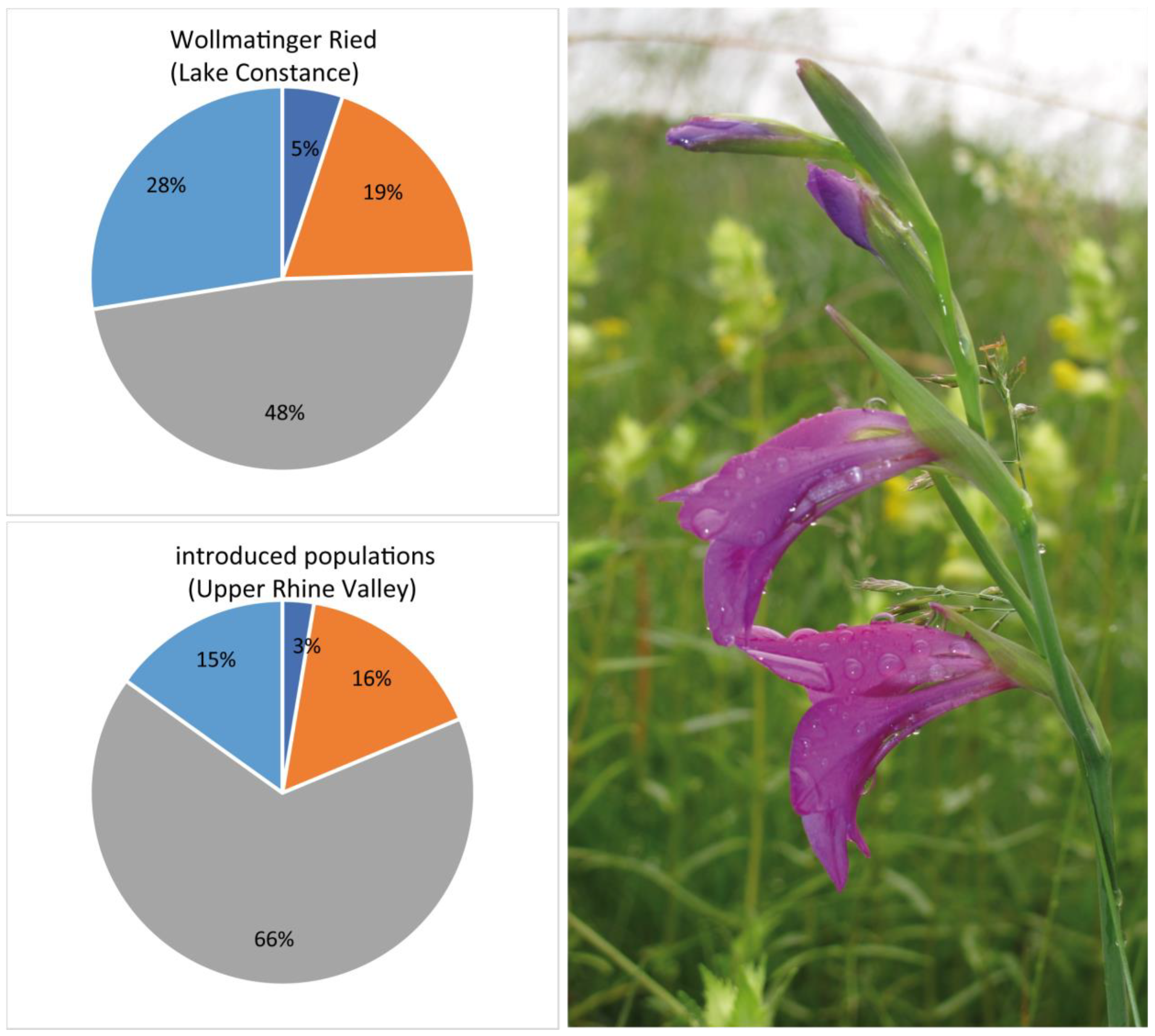
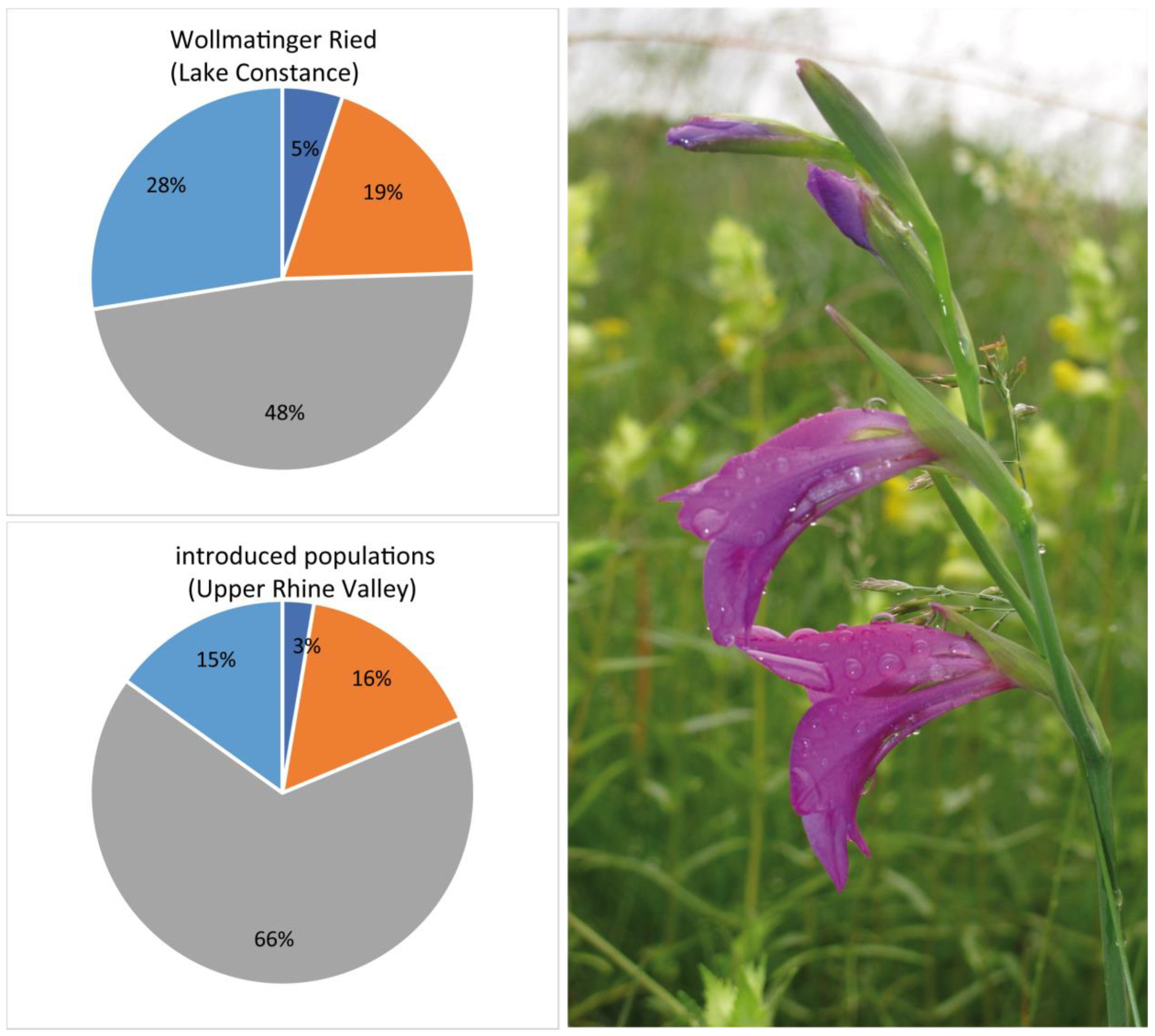
3.4. Genetic Assignment of Putatively Introduced Populations by Humans in Baden–Württemberg during the Last 30 Years
4. Discussion
4.1. Gladiolus in Central Europe Is a Reticulate Species Complex
4.2. Hybrids, Introgressed and Also Introduced Populations of G. palustris Should Be Considered Targets of Species Conservation Efforts
Supplementary Materials
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tscharntke, T.; Tylianakis, J.M.; Rand, T.A.; Didham, R.K.; Fahrig, L.; Batáry, P.; Bengtsson, J.; Clough, Y.; Crist, T.O.; Dormann, C.F.; et al. Landscape moderation of biodiversity patterns and processes—Eight hypotheses. Biol. Rev. 2012, 87, 661–685. [Google Scholar] [CrossRef] [PubMed]
- Steffen, W.; Richardson, K.; Rockström, J.; Cornell, S.E.; Fetzer, I.; Bennet, E.M.; Biggs, R.; Carpenter, S.R.; De Vries, W.; De Witt, C.A.; et al. Planetary boundaries: Guiding human development on a changing planet. Science 2015, 347, 1259855. [Google Scholar] [CrossRef] [PubMed]
- Thuiller, W.; Lavorel, S.; Araújo, M.B.; Sykes, M.T.; Prentice, I.C. Climate change threats to plant diversity in Europe. Proc. Natl. Acad. Sci. USA 2005, 102, 8245–8250. [Google Scholar] [CrossRef] [PubMed]
- Reed, D.H.; Frankham, R. Correlation between fitness and genetic diversity. Conserv. Biol. 2003, 17, 230–237. [Google Scholar] [CrossRef]
- Chan, W.Y.; Hoffmann, A.A.; van Oppen, M.J.H. Hybridization as a conservation management tool. Conserv. Lett. 2019, 12, e12652. [Google Scholar] [CrossRef]
- Abbott, R.; Albach, D.; Ansell, S.; Arntzen, J.W.; Baird, S.J.E.; Bierne, N.; Boughman, J.; Brelsford, A.; Buerkle, C.A.; Buggs, R.; et al. Hybridization and speciation. J. Evol. Biol. 2013, 26, 229–246. [Google Scholar] [CrossRef]
- Abbott, R.J. Plant speciation across environmental gradients and the occurrence and nature of hybrid zones. J. Syst. Evol. 2017, 55, 238–258. [Google Scholar] [CrossRef]
- Hohmann, N.; Koch, M.A. An Arabidopsis introgression zone studied at high spatio-temporal resolution: Interglacial and multiple genetic contact exemplified using whole nuclear and plastid genomes. BMC Genom. 2017, 18, 810. [Google Scholar] [CrossRef]
- Koch, M. Genetic differentiation and speciation in prealpine Cochlearia: Allohexaploid Cochlearia bavarica Vogt (Brassicaceae) compared to its diploid ancestor Cochlearia pyrenaica DC. in Germany and Austria. Plant Syst. Evol. 2002, 232, 35–49. [Google Scholar] [CrossRef]
- Pachschwöll, C.; Escobar García, P.; Winkler, M.; Schneeweiss, G.M.; Schönswetter, P. Polyploidisation and geographic differentiation drive diversification in a European high mountain plant group (Doronicum clusii aggregate, Asteraceae). PLoS ONE 2015, 10, e0118197. [Google Scholar] [CrossRef]
- Hebda, A.; Kempf, M.; Wachowiak, W.; Pluciński, B.; Kauzal, P.; Zwijacz-Kozica, T. Hybridization and introgression of native and foreign Sorbus tree species in unique environments of protected mountainous areas. AoB Plants 2021, 13, plaa070. [Google Scholar] [CrossRef] [PubMed]
- Wörz, A. Untersuchungen zu Abgrenzung, Verbreitung und Standortswahl von Anthriscus sylvestris (L.) Hoffm. ssp. stenophylla (Rouy & Camus) Briquet (Apiaceae). Bot. Jahrb. Syst. 1992, 114, 329–348. [Google Scholar]
- Nierbauer, K.U.; Kanz, B.; Zizka, G. The widespread naturalization of Nymphaea hybrids is masking the decline of wild-type Namphaea alba in Hesse, Germany. Flora 2014, 209, 122–130. [Google Scholar] [CrossRef]
- Arrigo, N.; Bétrisey, S.; Graf, L.; Bilat, J.; Gerber, E.; Kozlowski, G. Hybridization as a threat in climate relict Nuphar pumila (Nymphaeaceae). Biodivers. Conserv. 2016, 25, 1863–1877. [Google Scholar] [CrossRef]
- Allendorf, F.W.; Leary, R.F.; Spruell, P.; Wenburg, J.K. The problems with hybrids: Setting conservation guidelines. Trends Ecol. Evol. 2001, 16, 613–622. [Google Scholar] [CrossRef]
- Draper, D.; Laguna, E.; Marques, I. Demystifying negative connotations of hybridization for less biased conservation policies. Front. Ecol. Evol. 2021, 9, 637100. [Google Scholar] [CrossRef]
- Schmitt, B.; Fartmann, T.; Hölzel, N. Vergesellschaftung und Ökologie der Sumpf-Siegwurz (Gladiolus palustris) in Südbayern. Tuexenia 2010, 30, 105–127. [Google Scholar]
- Szczepaniak, M.; Kaminski, R.; Kuta, E.; Slomka, A.; Heise, W.; Cieslak, E. Natural hybridization between Gladiolus palustris and G. imbricatus inferred from morphological, molecular and reproductive evidence. Preslia 2016, 88, 137–161. [Google Scholar]
- Daco, L.; Maurice, T.; Muller, S.; Rossa, J.; Colling, G. Genetic status of the endangered plant species Gladiolus palustris in the western part of its distribution. Conserv. Genet. 2019, 20, 1339–1354. [Google Scholar] [CrossRef]
- Cieślak, E.; Szczepaniak, M.; Kamiński, R.; Heise, W. Stan zachowania krytycznie zagrożonego gatunku Gladiolus paluster (Iridaceae) w Polsce—Analiza zmienności genetycznej osobników w uprawie Ogrodu Botanicznego Uniwersytetu Wrocławskiego w kontekście prowadzonych działań ochronnych. Fragm. Florist. Geobot. Pol. 2014, 21, 49–66. [Google Scholar]
- Leitner, B.M. Genetic Structure and Pollination Biology of the FFH-Protected Gladiolus palustris GAUDIN (Iridaceae) in Salzburg and Adjacent Areas. Master’s Thesis, University Salzburg, Salzburg, Austria, 2018; pp. 1–106. [Google Scholar]
- Doyle, J.; Doyle, J. A rapid isolation procedure for small amounts of leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar] [CrossRef]
- Gong, W.; Chen, C.; Dobeš, C.; Fu, C.X.; Koch, M.A. Phylogeography of a living fossil: Pleistocene glaciations forced Ginkgo biloba L. (Ginkgoaceae) into two refuge areas in China with limited subsequent postglacial expansion. Mol. Phylogenet. Evol. 2008, 48, 1094–1105. [Google Scholar] [CrossRef]
- Vos, P.; Hogers, R.; Bleeker, M.; Reijans, M.; Lee Van De, T.; Hornes, M.; Frijters, A.; Pot, J.; Peleman, J.; Kuiper, M.; et al. AFLP: A new technique for DNA fingerprinting. Nucleic Acids Res. 1995, 23, 4407–4414. [Google Scholar] [CrossRef]
- Koch, M.A.; Winizuk, A.; Banzhaf, P.; Reichardt, J. Impact of climate change on the success of population support management and plant reintroduction at steep, exposed limestone outcrops in the German Swabain Jura. Perspect. Plant Ecol. Evol. Syst. 2021, 53, 125643. [Google Scholar] [CrossRef]
- Bonin, A.; Bellemain, E.; Eidesen, P.B.; Pompanon, F.; Brochmann, C.; Taberlet, P. How to track and assess genotyping errors in population genetics studies. Mol. Ecol. 2004, 13, 3261–3273. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Development Core Team; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Bryant, D.; Moulton, V. Neighbor-net: An agglomerative method for the construction of phylogenetic networks. Mol. Biol. Evol. 2004, 21, 255–265. [Google Scholar] [CrossRef]
- Huson, D.H. SplitsTree: Analyzing and visualizing evolutionary data. Bioinformatics 1998, 14, 68–73. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Molec. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Molec. Ecol. Notes 2007, 7, 574–578. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Earl, D.A.; von Holdt, B.M. Structure Harvester: A website and program for visualizing structure output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal component Analysis and heatmap. Nucleic Acid. Res. 2015, 43, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Guglielmo, F.; Poggio, L.; Tutino, S. DNA Barcoding of Land Plant Species in Aosta Valley (Northwest Italy). Available online: https://www.ncbi.nlm.nih.gov/nuccore/MF543717 (accessed on 4 March 2023).
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2005, 32, 268–274. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslonko, M.; van der Mark, P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Wörz, A.; Voggesberger, M.; Abrahamczyk, S.; Krause, C.; Bildstein, U.; Thiv, M. Aktuelle Verbreitungskarten der Farn- und Blütenpflanzen Baden-Württembergs. Available online: http://www.flora.naturkundemuseum-bw.de (accessed on 5 January 2023).
- Koch, M.A.; Dobes, C.; Mitchell-Olds, T. Multiple hybrid formation in natural populations: Concerted evolution of the internal transcribed spacer of nuclear ribosomal DNA (ITS) in North American Arabis divaricarpa (Brassicaceae). Mol. Biol. Evol. 2003, 20, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.P. Gladiolus L. Flora Europaea, Alismataceae to Orchidaceae (Monocotyledones); Tutin, T.G., Heywood, V.H., Burges, N.A., Moore, D.M., Valentine, D.H., Walters, S.M., Webb, D.A., Eds.; Cambridge University Press: Cambridge, UK, 1980; Volume 5, pp. 101–102. [Google Scholar]
- Van Raamsdonk, L.W.D.; de Vries, T. Biosystematic studies in European species of Gladiolus (Iridaceae). Plant Syst. Evol. 1989, 165, 189–198. [Google Scholar] [CrossRef]
- Bohling, J.H. Strategies to address the conservation threats posed by hybridization and genetic introgression. Biol. Conserv. 2016, 203, 321–327. [Google Scholar] [CrossRef]
- Carrió, E.; Güemes, J. Can the problem of hybridization in threatened species be evaluated using a fieldwork research? A case study in snapdragons. J. Nat. Conserv. 2019, 48, 47–53. [Google Scholar] [CrossRef]
- Jackiw, R.N.; Mandil, G.; Hager, H.A. A framework to guide the conservation of species hybrids based on ethical and ecological considerations. Conserv. Biol. 2015, 29, 1040–1051. [Google Scholar] [CrossRef] [PubMed]
- Schnittler, M.; Günther, K.-F. Central European vascular plants requiring priority conservation measures—An analysis from national Red Lists and distribution maps. Biodiv. Conserv. 1999, 8, 891–925. [Google Scholar] [CrossRef]
- Bilz, M. Gladiolus palustris—The IUCN Red List of Threatened Species; Version 2015-3. Available online: http://www.iucnredlist.org (accessed on 13 October 2022).
- Bilz, M.; Kell, S.P.; Maxted, N.; Lansdown, R.V. European Red List of Vascular Plants; Publications Office of the European Union: Luxembourg, 2011; pp. 1–130. [Google Scholar] [CrossRef]
- Cantor, M.; Tolety, J. Gladiolus. In Wild Crop Relatives: Genomic and Breeding Resources, Plantation and Ornamental Crops; Kole, C., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 133–159. [Google Scholar]
- Goldblatt, P.; Manning, J.C.; Bernhardt, P. Radiation of pollination systems in Gladiolus (Iridaceae: Crocoideae) in Southern Africa. Ann. Miss. Bot. Gard. 2001, 88, 713–734. [Google Scholar] [CrossRef]
- Janes, J.K.; Hamilton, J.A. Mixing it up: The role of hybridization in forest management and conservation under climate change. Forests 2017, 8, 237. [Google Scholar] [CrossRef]
- Van Wyk, A.M.; Dalton, D.L.; Hoban, S.; Bruford, M.W.; Russo, I.M.; Birss, C.; Grobler, P.; van Vuuren, B.J.; Kotzé, A. Quantitative evaluation of hybridization and the impact on biodiversity conservation. Ecol. Evol. 2016, 7, 320–330. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N/n | Expected Heterozygosity | Gene Diversity | No. Polymorphic Loci | No. Rare Allels (<5%) | |
|---|---|---|---|---|---|
| Group 1 | 20/40 | 0.227 (0.159) | 0.073 (0.036) | 100 | 0.92 (0.91) |
| [Group 1 | 22/42 | 0.206 (0.156) | 0.077 (0.038) | 117 | 1.07 (1.07)] |
| Group 2 * | 9/23 | 0.255 (0.159) | 0.067 (0.034) | 82 | 0.90 (0.97) |
| [Group 2 | 11/25 | 0.201 (0.149) | 0.088 (0.044) | 138 | 1.73 (4.21)] |
| Group 3 * | 7/17 | 0.280 (0.152) | 0.081 (0.042) | 90 | 2.33 (1.41) |
| [Group 3 | 9/20 | 0.229 (0.149) | 0.106 (0.054) | 145 | 2.33 (1.36)] |
| Source of Variation | d.f. | Sum of Squares | Variance Components | Percentage of Variation | |
|---|---|---|---|---|---|
| (a) | |||||
| reduced | among populations | 2 | 81.034 | 1.171 (Va) | 9.34 |
| non-reduced | 2 | 90.838 | 1.158 (Va) | 7.84 | |
| reduced | within populations | 77 | 875.341 | 11.368 (Vb) | 90.66 |
| non-reduced | 84 | 1143.231 | 13.609 (Vb) | 92.16 | |
| (b) | |||||
| reduced | among groups | 2 | 81.034 | 0.858 (Va) | 6.87 |
| non-reduced | 2 | 90.838 | 0.727 (Va) | 4.94 | |
| reduced | among populations | 33 | 488.930 | 2.858 (Vb) | 22.87 |
| non-reduced | within groups | 39 | 721.670 | 4.624 (Vb) | 31.42 |
| reduced | within populations | 44 | 386.441 | 8.782 (Vc) | 70.26 |
| non-reduced | 45 | 421.561 | 9.368 (Vc) | 63.64 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koch, M.A. The Diverse Reticulate Genetic Set-Up of Endangered Gladiolus palustris in Southern Germany Has Consequences for the Development of Conservation Strategies. Diversity 2023, 15, 1068. https://doi.org/10.3390/d15101068
Koch MA. The Diverse Reticulate Genetic Set-Up of Endangered Gladiolus palustris in Southern Germany Has Consequences for the Development of Conservation Strategies. Diversity. 2023; 15(10):1068. https://doi.org/10.3390/d15101068
Chicago/Turabian StyleKoch, Marcus A. 2023. "The Diverse Reticulate Genetic Set-Up of Endangered Gladiolus palustris in Southern Germany Has Consequences for the Development of Conservation Strategies" Diversity 15, no. 10: 1068. https://doi.org/10.3390/d15101068
APA StyleKoch, M. A. (2023). The Diverse Reticulate Genetic Set-Up of Endangered Gladiolus palustris in Southern Germany Has Consequences for the Development of Conservation Strategies. Diversity, 15(10), 1068. https://doi.org/10.3390/d15101068