
A Tale of Native American Whole-Genome Sequencing and Other Technologies

 ,
,  , ,
, ,  and
and 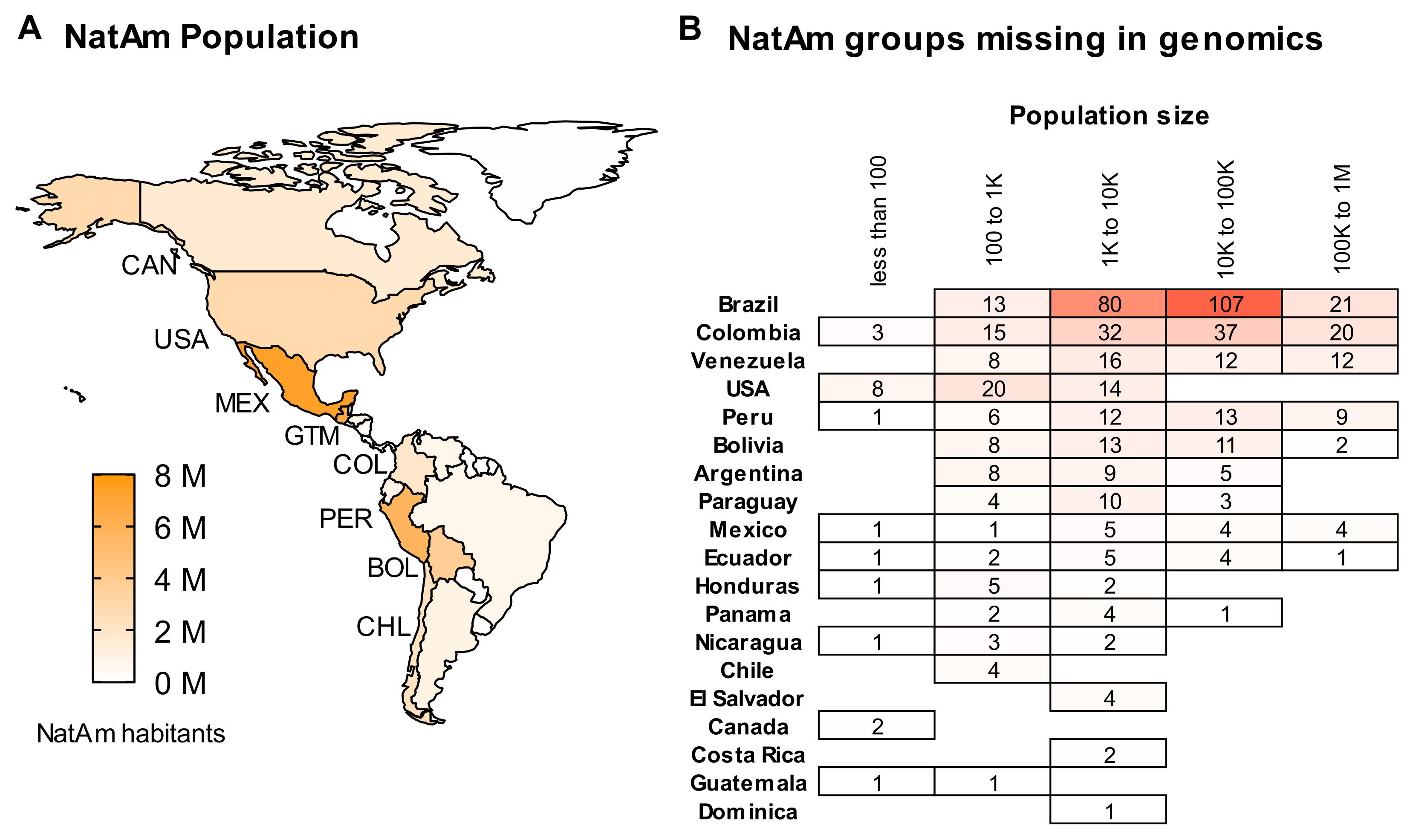{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
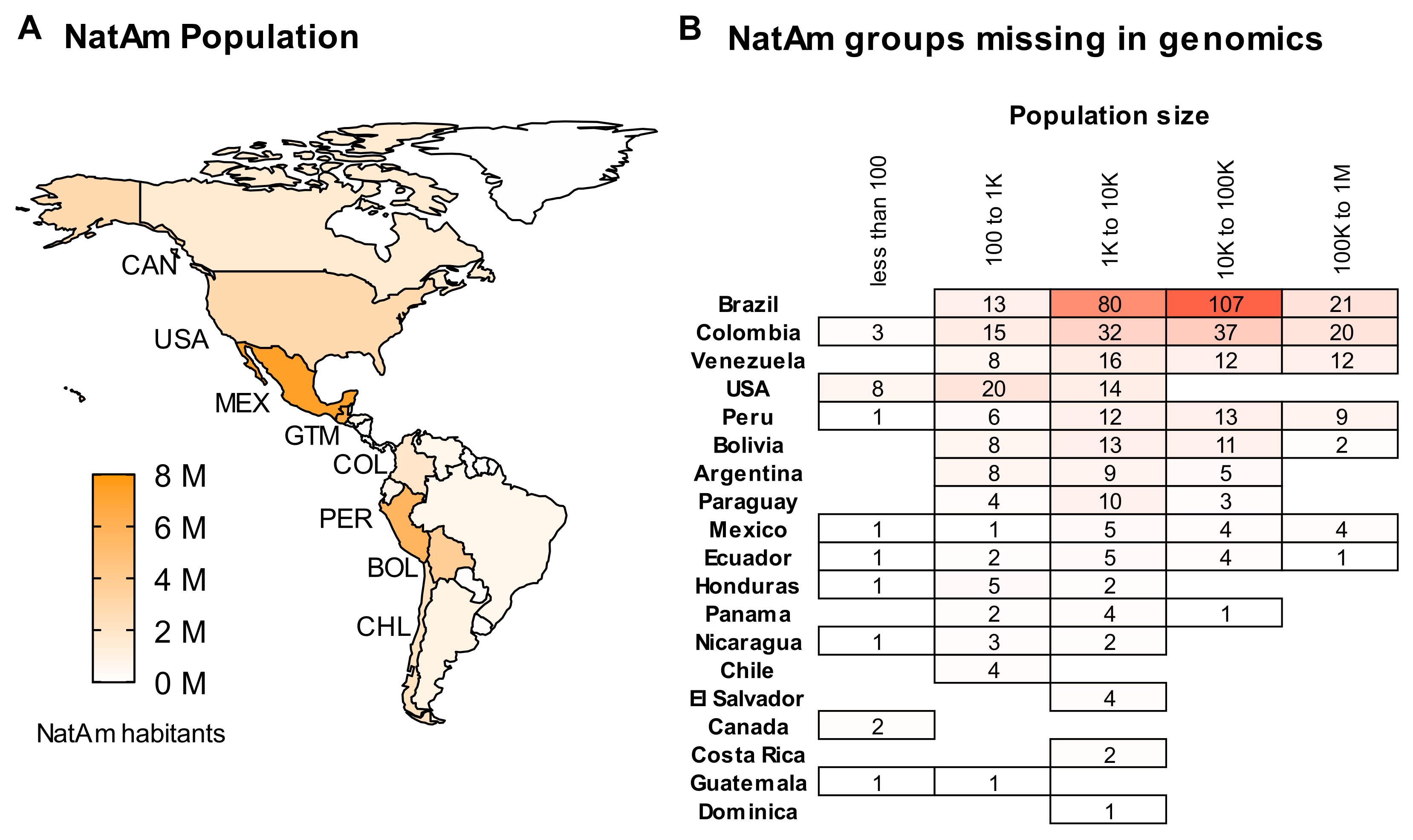
2. A Summary of Native American Populations
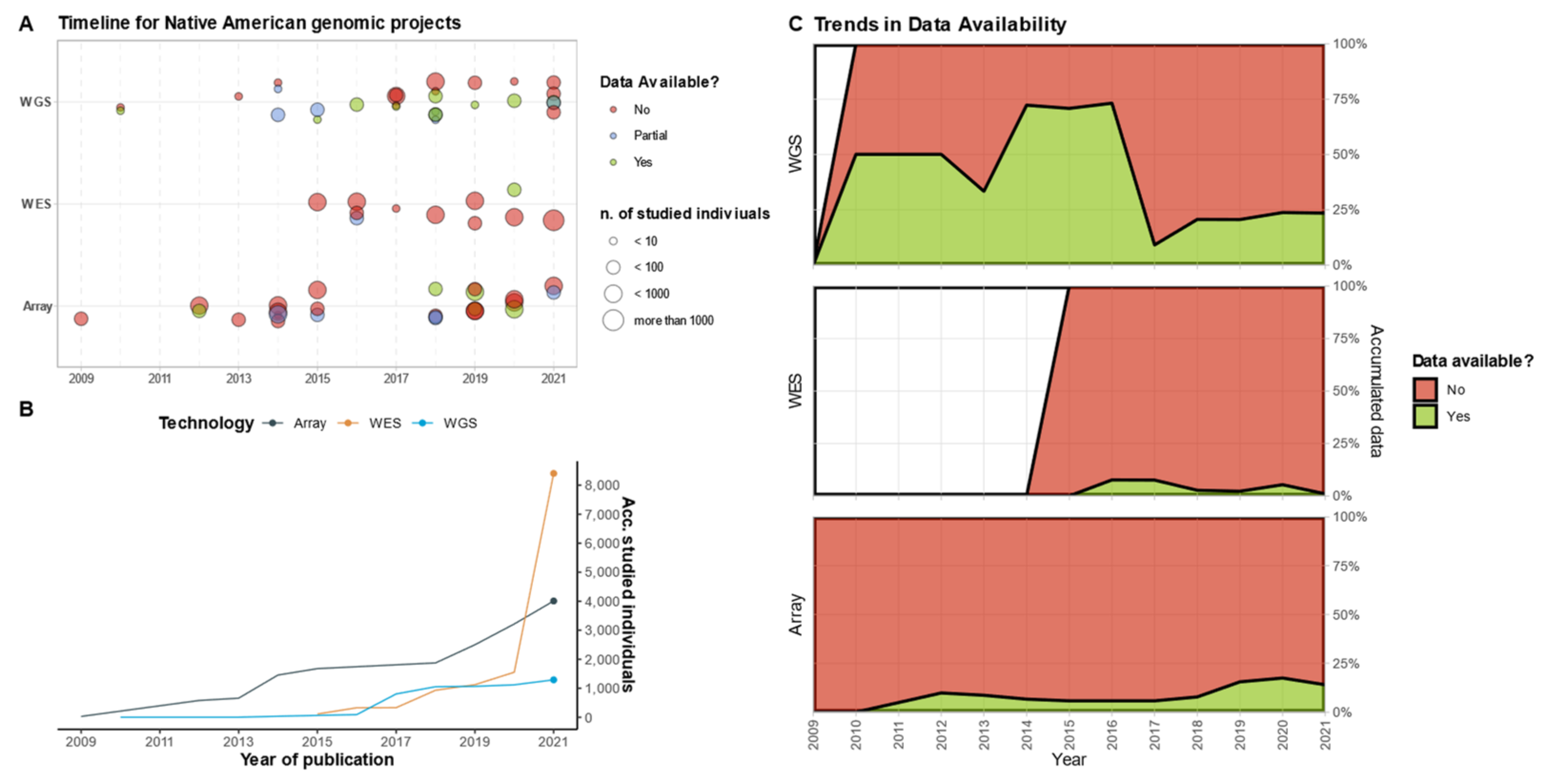
3. The Technology behind Population Genomics
4. To Sequence or Not to Sequence?
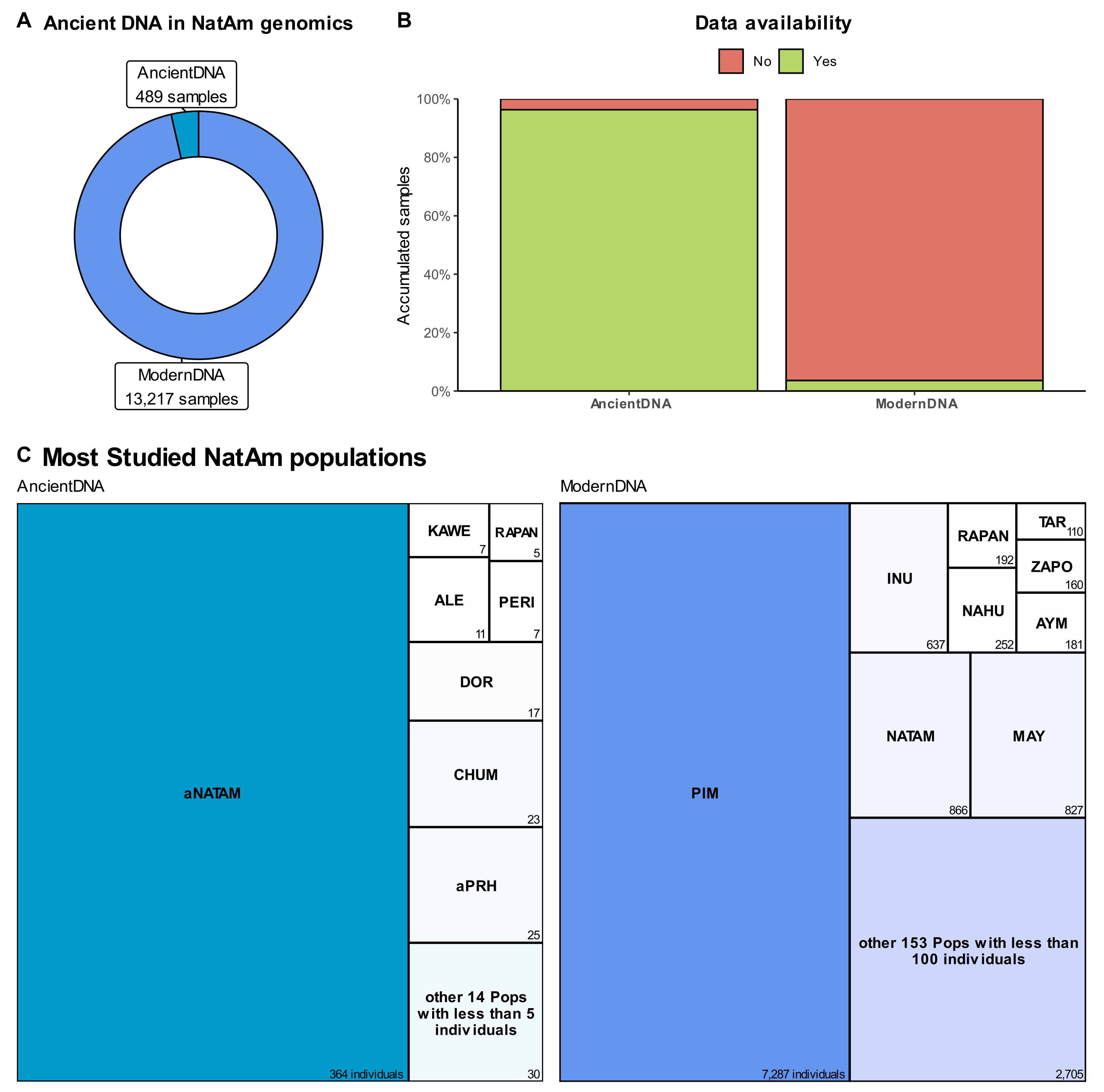
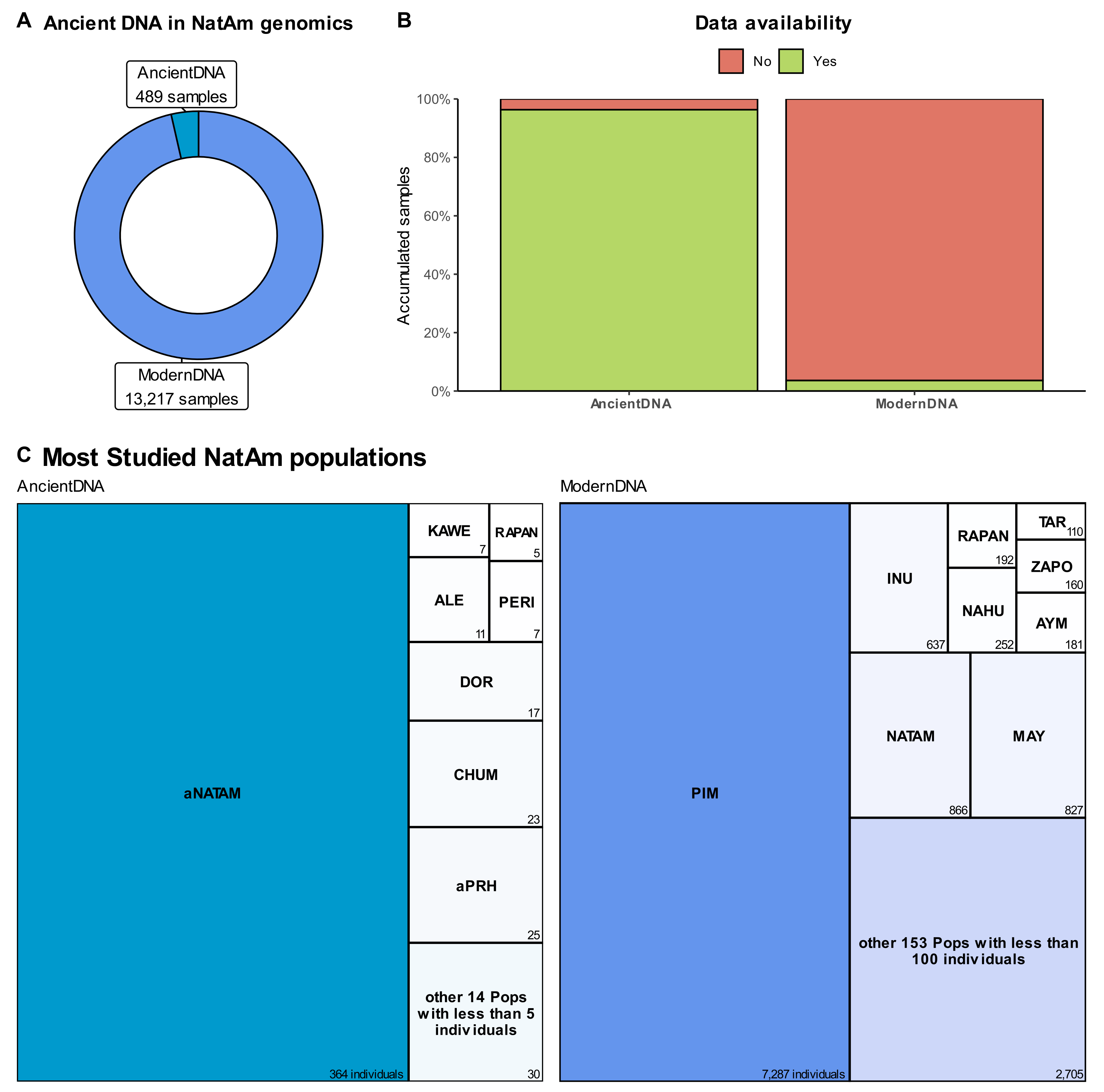
5. Sequencing, by the Numbers
6. About Data Availability and the Missing NatAm Groups
7. Concluding Remarks
8. Methods
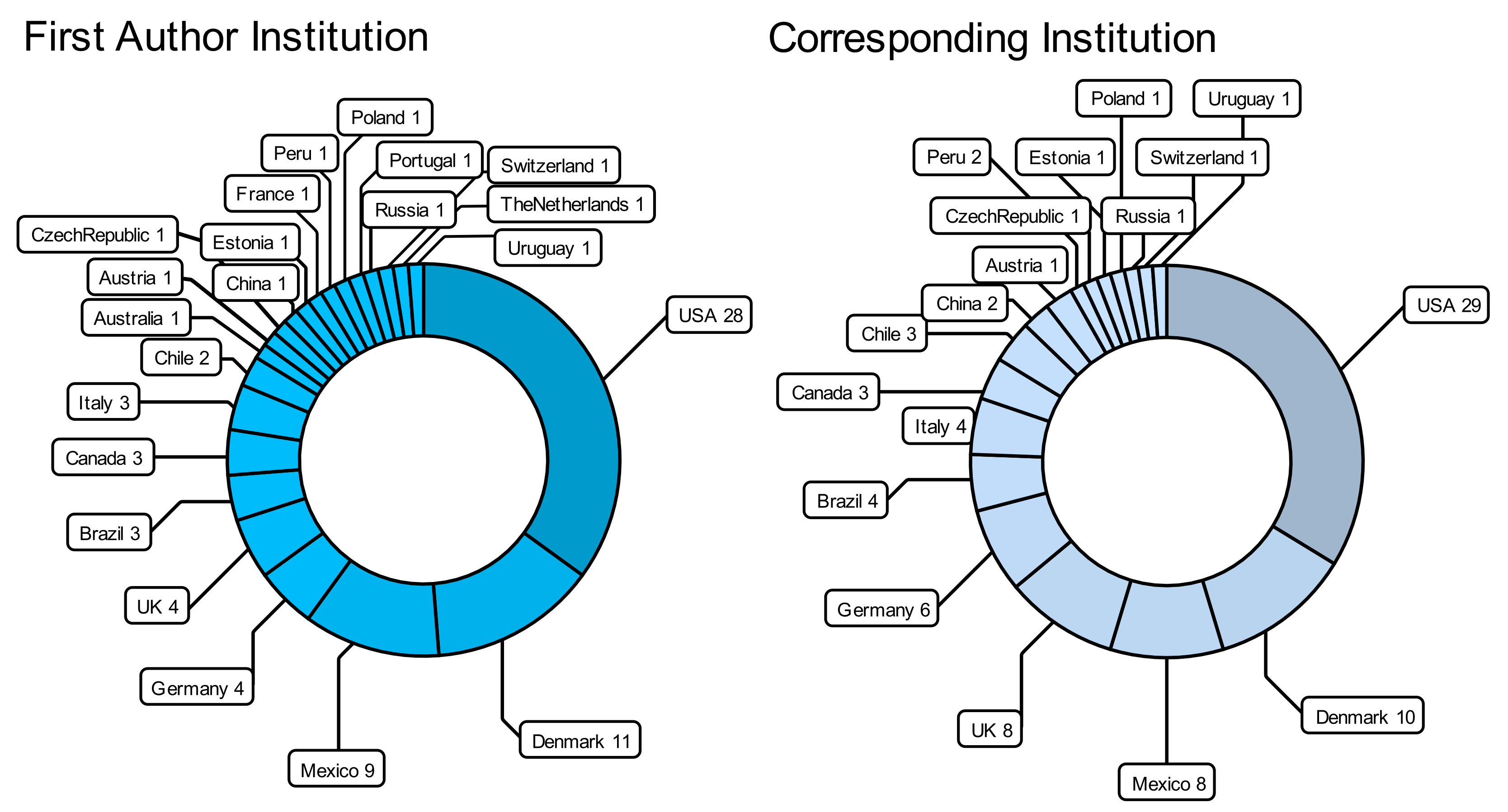
8.1. Data Collection
8.2. Census Information on NatAm Home Countries
8.3. Data Handling and Visualization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [PubMed]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Ballouz, S.; Dobin, A.; Gillis, J.A. Is it time to change the reference genome? Genome Biol. 2019, 20, 159. [Google Scholar] [CrossRef] [PubMed]
- Bergström, A.; McCarthy, S.A.; Hui, R.; Almarri, M.A.; Ayub, Q.; Danecek, P.; Chen, Y.; Felkel, S.; Hallast, P.; Kamm, J.; et al. Insights into human genetic variation and population history from 929 diverse genomes. Science 2020, 367, eaay5012. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Collins, R.L.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alföldi, J.; Francioli, L.C.; Khera, A.V.; Lowther, C.; Gauthier, L.D.; Wang, H.; et al. A structural variation reference for medical and population genetics. Nature 2020, 581, 444–451. [Google Scholar] [CrossRef]
- Need, A.C.; Goldstein, D.B. Next generation disparities in human genomics: Concerns and remedies. Trends Genet. 2009, 25, 489–494. [Google Scholar] [CrossRef]
- Bustamante, C.D.; De La Vega, F.M.; Burchard, E.G. Genomics for the world. Nature 2011, 475, 163–165. [Google Scholar] [CrossRef]
- Popejoy, A.B.; Fullerton, S.M. Genomics is failing on diversity. Nature 2016, 538, 161–164. [Google Scholar] [CrossRef]
- Sirugo, G.; Williams, S.M.; Tishkoff, S.A. The Missing Diversity in Human Genetic Studies. Cell 2019, 177, 26–31. [Google Scholar] [CrossRef]
- Caron, N.R.; Chongo, M.; Hudson, M.; Arbour, L.; Wasserman, W.W.; Robertson, S.; Correard, S.; Wilcox, P. Indigenous Genomic Databases: Pragmatic Considerations and Cultural Contexts. Front. Public Health 2020, 8, 11. [Google Scholar] [CrossRef]
- Guglielmi, G. Facing up to injustice in genome science. Nature 2019, 568, 290–293. [Google Scholar] [CrossRef]
- Pedersen, M.W.; Ruter, A.; Schweger, C.; Friebe, H.; Staff, R.A.; Kjeldsen, K.K.; Mendoza, M.L.Z.; Beaudoin, A.B.; Zutter, C.; Larsen, N.K.; et al. Postglacial viability and colonization in North America’s ice-free corridor. Nature 2016, 537, 45–49. [Google Scholar] [CrossRef]
- Willerslev, E.; Meltzer, D.J. Peopling of the Americas as inferred from ancient genomics. Nature 2021, 594, 356–364. [Google Scholar] [CrossRef]
- Skoglund, P.; Reich, D. A genomic view of the peopling of the Americas. Curr. Opin. Genet. Dev. 2016, 41, 27–35. [Google Scholar] [CrossRef]
- Potter, B.A.; Baichtal, J.F.; Beaudoin, A.B.; Fehren-Schmitz, L.; Haynes, C.V.; Holliday, V.T.; Holmes, C.E.; Ives, J.W.; Kelly, R.L.; Llamas, B.; et al. Current evidence allows multiple models for the peopling of the Americas. Sci. Adv. 2018, 4, eaat5473. [Google Scholar] [CrossRef]
- Bennett, M.R.; Bustos, D.; Pigati, J.S.; Springer, K.B.; Urban, T.M.; Holliday, V.T.; Reynolds, S.C.; Budka, M.; Honke, J.S.; Hudson, A.M.; et al. Evidence of humans in North America during the Last Glacial Maximum. Science 2021, 373, 1528–1531. [Google Scholar] [CrossRef]
- Raghavan, M.; Steinrücken, M.; Harris, K.; Schiffels, S.; Rasmussen, S.; DeGiorgio, M.; Albrechtsen, A.; Valdiosera, C.; Ávila-Arcos, M.C.; Malaspinas, A.; et al. Genomic evidence for the Pleistocene and recent population history of Native Americans. Science 2015, 349, aab3884. [Google Scholar] [CrossRef]
- Scheib, C.L.; Li, H.; Desai, T.; Link, V.; Kendall, C.; Dewar, G.; Griffith, P.W.; Morseburg, A.; Johnson, J.R.; Potter, A.; et al. Ancient human parallel lineages within North America contributed to a coastal expansion. Science 2018, 360, 1024–1027. [Google Scholar] [CrossRef]
- Bolnick, D.A.; Raff, J.A.; Springs, L.C.; Reynolds, A.W.; Miró-Herrans, A.T. Native American Genomics and Population Histories. Annu. Rev. Anthropol. 2016, 45, 319–340. [Google Scholar] [CrossRef]
- United Nations, Department of Economic and Social Affairs, Population Division. World Population Prospects 2019; UN: New York, NY, USA, 2019; Volume II. [Google Scholar]
- Instituto Nacional de Estadística y Geografía. Censo de Población y Vivienda 2020. 2021. Available online: https://www.inegi.org.mx/programas/ccpv/2020/#Tabulados (accessed on 8 March 2022).
- Instituto Nacional de Estadística. Portal de Resultados del Censo 2018. Available online: https://www.censopoblacion.gt/explorador (accessed on 8 March 2022).
- Base de Datos de Pueblos Indígenas u Originarios. Lista de Pueblos Indígenas u Originarios. Available online: https://bdpi.cultura.gob.pe/pueblos-indigenas (accessed on 8 March 2022).
- Instituto Nacional de Estadística. Censos. INE. Available online: https://www.ine.gob.bo/index.php/censos-y-banco-de-datos/censos/ (accessed on 8 March 2022).
- Norris, T.; Vines, P.L.; Hoeffel, E.M. The American Indian and Alaska Native Population: 2010; US Department of Commerce, Economics and Statistics Administration, US Census Bureau: Suitland, MD, USA, 2010; Volume 21. [Google Scholar]
- Censo 2017. 2017. Available online: https://www.censo2017.cl/descargas/home/sintesis-de-resultados-censo2017.pdf (accessed on 17 May 2022).
- Departamento Administrativo Nacional de Estadística. Grupos Étnicos Información Técnica. 2018. Available online: https://www.dane.gov.co/index.php/estadisticas-por-tema/demografia-y-poblacion/grupos-etnicos/informacion-tecnica (accessed on 8 March 2022).
- Government of Canada SC. Aboriginal Population Profile, 2016 Census—Canada [Country]. 2018. Available online: https://www12.statcan.gc.ca/census-recensement/2016/dp-pd/abpopprof/details/page.cfm?Lang=E&Geo1=PR&Code1=01&Data=Count&SearchText=Canada&SearchType=Begins&B1=Aboriginal%20peoples&C1=All&SEXID=1&AGEID=1&RESGEOID=1 (accessed on 8 March 2022).
- INDEC (Instituto Nacional de Estadística y Censos de la República Argentina). Pueblos Originarios. 2010. Available online: https://www.indec.gob.ar/indec/web/Nivel4-Tema-2–21–99 (accessed on 8 March 2022).
- Silverio Chisaguano, M. La Población Indígena del Ecuador; Instituto Nacional de Estadística y Censos (INEC): Loja, Ecuador, 2006; p. 39. [Google Scholar]
- Gerencia General de Estadísticas Demográficas. Resultados Población Indígena. XIV Censo de Población y Vivienda 2011; Instituto Nacional de Estadística: Caracas, Venezuela, 2011; p. 42. Available online: http://www.ine.gov.ve/documentos/Demografia/CensodePoblacionyVivienda/pdf/ResultadosBasicos.pdf (accessed on 9 March 2022).
- Quadro Geral dos Povos—Povos Indígenas No Brasil. 2021. Available online: https://pib.socioambiental.org/pt/Quadro_Geral_dos_Povos (accessed on 9 March 2022).
- Instituto Nacional de Estadística de Honduras. Tomo 6. Grupos Poblacionales. 2015. Available online: https://www.ine.gob.hn/publicaciones/Censos/Censo_2013/06Tomo-VI-Grupos-Poblacionales/cuadros.html (accessed on 9 March 2022).
- Davis, E. Diagnóstico de la Población Indígena de Panamá con Base en los Censos de Población y Vivienda de 2010; Instituto Nacional de Estadística y Censo (INEC): Panama City, Panama, 2010; p. 186. Available online: https://inec.gob.pa/archivos/P6571INDIGENA_FINAL_FINAL.pdf (accessed on 9 March 2022).
- Oficina Pueblos Indígenas. Nota técnica de país sobre cuestiones de los pueblos indígenas. República de Nicaragua: Centro para la Autonomía y Desarrollo de los Pueblos Indígenas (CADPI). 2017. Available online: https://www.ifad.org/documents/38714170/40258424/nicaragua_ctn_s.pdf (accessed on 19 May 2022).
- Otazú, N.; Bazán, R.; Leiva, V.M. Censo de Comunidades de los Pueblos Indígenas Resultados Finales 2012; Dirección General de Estadística, Encuestas y Censos (DGEEC): Asuncion, Paraguay, 2012; p. 140. Available online: https://www.ine.gov.py/Publicaciones/Biblioteca/documento/c3c9_Censo%20de%20Comunidades%20de%20los%20Pueblos%20Indigenas%20Resultados%20Finales%202012.pdf (accessed on 9 March 2022).
- Instituto Nacional de Estadística y Censos. Costa Rica: Población Indígena por Pertenencia a un Pueblo Indígena, Según Provincia y Sexo. 2011. Available online: https://www.inec.cr/sites/default/files/documentos/poblacion/estadisticas/resultados/resocialcenso2011–03.xls.xls (accessed on 22 March 2022).
- International Work Group for Indigenous Affairs (IWGIA). Indigenous Peoples in Guyana. Available online: https://iwgia.org/en/guyana.html (accessed on 9 March 2022).
- Rodriguez, P.; Justo, C.; Miguel, C.; Olivera, J.; Martino, D. Población Indígena en Uruguay y su Vínculo con el Bosque. Proyecto REDD+ Uruguay; Ministerio de Ganadería, Agricultura y Pesca-Ministerio de Vivienda, Ordenamiento Territorial y Medio Ambiente: Montevideo, Uruguay, 2020; p. 47. Available online: https://www.gub.uy/ministerio-ganaderia-agricultura-pesca/sites/ministerio-ganaderia-agricultura-pesca/files/documentos/publicaciones/1.%20Informe_PI_y_BN.pdf (accessed on 9 March 2022).
- Grønlands Statistik. Available online: https://stat.gl/dialog/topmain.asp?lang=da&subject=Befolkning&sc=BE (accessed on 9 March 2022).
- The Statistical Institute of Belize. Belize Population and Housing Census (Country Report 2010); The Statistical Institute of Belize: Belmopan, Belize, 2013; p. 176. Available online: http://sib.org.bz/wp-content/uploads/2010_Census_Report.pdf (accessed on 20 May 2022).
- General Bureau of Statistics. Mosaic of the Surinamese People. 2019. Available online: https://statistics-suriname.org/en/mosaic_of-the-surinamese-people/ (accessed on 20 May 2022).
- Censo de Población y Vivienda 2007 (Población). El Salvador: Dirección General de Estadística y Censos (DIGESTYC). 2007. Available online: http://www.digestyc.gob.sv/index.php/temas/des/poblacion-y-estadisticas-demograficas/censo-depoblacion-y-vivienda/poblacion-censos.html (accessed on 9 March 2022).
- International Work Group for Indigenous Affairs (IWGIA). Indigenous Peoples in French Guiana. Available online: https://iwgia.org/en/french-guiana.html (accessed on 9 March 2022).
- Population and Housing Census 2012. St. Vincent and the Grenadines: Statistical Office, Ministry of Finance, Government of St Vincent and the Grenadines. 2012. Available online: https://redatam.org/binsvg/RpWebEngine.exe/Portal?BASE=SVG2012 (accessed on 20 May 2022).
- Ethnic Groups by Sex 1991 2001 and 2011. Commonweal of Dominica: Central Statistics Office of Dominica. Available online: https://stats.gov.dm/subjects/demographic-statistics/ethnic-groups-by-sex-1991-2001-and-2011/ (accessed on 9 March 2022).
- Government of St. Lucia. The 2010 Population and Housing Census of St. Lucia; The Central Statistics Office, Government of St. Lucia: Castries, Saint Lucia, 2010. Available online: https://redatam.org/binlca/RpWebEngine.exe/Portal?BASE=PHC2010C&lang=ENG (accessed on 9 March 2022).
- Stephens, Z.D.; Lee, S.Y.; Faghri, F.; Campbell, R.H.; Zhai, C.; Efron, M.J.; Iyer, R.; Schatz, M.C.; Sinha, S.; Robinson, G.E. Big Data: Astronomical or Genomical? PLoS Biol. 2015, 13, e1002195. [Google Scholar] [CrossRef]
- Ng, S.B.; Turner, E.H.; Robertson, P.D.; Flygare, S.D.; Bigham, A.W.; Lee, C.; Shaffer, T.; Wong, M.; Bhattacharjee, A.; Eichler, E.E.; et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009, 461, 272–276. [Google Scholar] [CrossRef]
- Udpa, N.; Ronen, R.; Zhou, D.; Liang, J.; Stobdan, T.; Appenzeller, O.; Yin, Y.; Du, Y.; Guo, L.; Cao, R.; et al. Whole genome sequencing of Ethiopian highlanders reveals conserved hypoxia tolerance genes. Genome Biol. 2014, 15, R36. [Google Scholar] [CrossRef]
- Francioli, L.C.; Menelaou, A.; Pulit, S.L.; Van Dijk, F.; Palamara, P.F.; Elbers, C.C.; Neerincx, P.B.T.; Ye, K.; Guryev, V.; The Genome of the Netherlands Consortium; et al. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat. Genet. 2014, 46, 818–825. [Google Scholar]
- Gilissen, C.; Hehir-Kwa, J.Y.; Thung, D.T.; van de Vorst, M.; van Bon, B.W.M.; Willemsen, M.H.; Kwint, M.; Janssen, I.M.; Hoischen, A.; Schenck, A.; et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014, 511, 344–347. [Google Scholar] [CrossRef]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef]
- ENCODE Project Consortium; Dunham, I.; Birney, E.; Lajoie, B.; Sanyal, A.; Dong, X.; Greven, M.; Lin, X.; Wang, J.; Whitfield, T.W.; et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Gresham, D.; Dunham, M.J.; Botstein, D. Comparing whole genomes using DNA microarrays. Nat. Rev. Genet. 2008, 9, 291–302. [Google Scholar] [CrossRef]
- Foo, J.N.; Liu, J.J.; Tan, E.K. Whole-genome and whole-exome sequencing in neurological diseases. Nat. Rev. Neurol. 2012, 8, 508–517. [Google Scholar] [CrossRef]
- Poulsen, J.B.; Lescai, F.; Grove, J.; Bækvad-Hansen, M.; Christiansen, M.; Hagen, C.M.; Maller, J.; Stevens, C.; Li, S.; Li, Q.; et al. High-Quality Exome Sequencing of Whole-Genome Amplified Neonatal Dried Blood Spot DNA. PLoS ONE 2016, 11, e0153253. [Google Scholar] [CrossRef]
- Gonzalez-Covarrubias, V.; Morales-Franco, M.; Cruz-Correa, O.F.; Martínez-Hernández, A.; García-Ortíz, H.; Barajas-Olmos, F.; Genis-Mendoza, A.D.; Martínez-Magaña, J.J.; Nicolini, H.; Orozco, L.; et al. Variation in Actionable Pharmacogenetic Markers in Natives and Mestizos From Mexico. Front. Pharmacol. 2019, 10, 1169. [Google Scholar] [CrossRef] [PubMed]
- Rustagi, N.; Zhou, A.; Watkins, W.S.; Gedvilaite, E.; Wang, S.; Ramesh, N.; Muzny, D.; Gibbs, R.A.; Jorde, L.B.; Yu, F.; et al. Extremely low-coverage whole genome sequencing in South Asians captures population genomics information. BMC Genom. 2017, 18, 396. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Kaufmann, A.; Chong, A.Y.; Cortés, A.; Quinto-Cortés, C.D.; Fernandez-Valverde, S.L.; Ferreyra-Reyes, L.; Cruz-Hervert, L.P.; Medina-Muñoz, S.G.; Sohail, M.; Palma-Martinez, M.J.; et al. Imputation Performance in Latin American Populations: Improving Rare Variants Representation with the Inclusion of Native American Genomes. Front. Genet. 2022, 12, 719791. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.R.; Bick, D.; Belmont, J.W.; Taylor, S.L.; Ashley, E.; Dimmock, D.; Jobanputra, V.; Kearney, H.M.; Kulkarni, S.; Rehm, H. The Medical Genome Initiative: Moving whole-genome sequencing for rare disease diagnosis to the clinic. Genome Med. 2020, 12, 48. [Google Scholar] [CrossRef]
- Bick, D.; Fraser, P.C.; Gutzeit, M.F.; Harris, J.M.; Hambuch, T.M.; Helbling, D.C.; Jacob, H.J.; Kersten, J.N.; Leuthner, S.R.; May, T.; et al. Successful Application of Whole Genome Sequencing in a Medical Genetics Clinic. J. Pediatr. Genet. 2017, 6, 61–76. [Google Scholar]
- Cortés-Ciriano, I.; Lee, J.J.K.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; Gordenin, D.; Klimczak, L.J.; Zhang, C.-Z.; Pellman, D.S.; et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat. Genet. 2020, 52, 331–341. [Google Scholar] [CrossRef]
- Dong, Z.; Xie, W.; Chen, H.; Xu, J.; Wang, H.; Li, Y.; Wang, J.; Chen, F.; Choy, K.W.; Jiang, H. Copy-Number Variants Detection by Low-Pass Whole-Genome Sequencing. Curr. Protoc. Hum. Genet. 2017, 94, 8.17.1–8.17.16. [Google Scholar] [CrossRef]
- Kosugi, S.; Momozawa, Y.; Liu, X.; Terao, C.; Kubo, M.; Kamatani, Y. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 2019, 20, 117. [Google Scholar] [CrossRef]
- Lippert, C.; Sabatini, R.; Maher, M.C.; Kang, E.Y.; Lee, S.; Arikan, O.; Harley, A.; Bernal, A.; Garst, P.; Lavrenko, V.; et al. Identification of individuals by trait prediction using whole-genome sequencing data. Proc. Natl. Acad. Sci. USA 2017, 114, 10166–10171. [Google Scholar] [CrossRef]
- Nakagawa, H.; Fujita, M. Whole genome sequencing analysis for cancer genomics and precision medicine. Cancer Sci. 2018, 109, 513–522. [Google Scholar] [CrossRef]
- Nielsen, R.; Akey, J.M.; Jakobsson, M.; Pritchard, J.K.; Tishkoff, S.; Willerslev, E. Tracing the peopling of the world through genomics. Nature 2017, 541, 302–310. [Google Scholar] [CrossRef]
- Aguilar-Ordoñez, I.; Pérez-Villatoro, F.; García-Ortiz, H.; Barajas-Olmos, F.; Ballesteros-Villascán, J.; González-Buenfil, R.; Fresno, C.; Garcíarrubio, A.; Fernández-López, J.C.; Tovar, H.; et al. Whole genome variation in 27 Mexican indigenous populations, demographic and biomedical insights. PLoS ONE 2021, 16, e0249773. [Google Scholar] [CrossRef]
- Bongers, J.L.; Nakatsuka, N.; O’Shea, C.; Harper, T.K.; Tantaleán, H.; Stanish, C.; Fehren-Schmitz, L. Integration of ancient DNA with transdisciplinary dataset finds strong support for Inca resettlement in the south Peruvian coast. Proc. Natl. Acad. Sci. USA 2020, 117, 18359–18368. [Google Scholar] [CrossRef]
- Day, S.E.; Muller, Y.L.; Koroglu, C.; Kobes, S.; Wiedrich, K.; Mahkee, D.; Baier, L.J. Exome Sequencing of 21 Bardet-Biedl Syndrome (BBS) Genes to Identify Obesity Variants in 6,851 American Indians. Obesity 2021, 29, 748–754. [Google Scholar] [CrossRef]
- Spangenberg, L.; Fariello, M.I.; Arce, D.; Illanes, G.; Greif, G.; Shin, J.-Y.; Yoo, S.-K.; Seo, J.-S.; Robello, C.; Kim, C.; et al. Indigenous Ancestry and Admixture in the Uruguayan Population. Front. Genet. 2021, 12, 1818. [Google Scholar] [CrossRef]
- García-Ortiz, H.; Barajas-Olmos, F.; Contreras-Cubas, C.; Cid-Soto, M.Á.; Córdova, E.J.; Centeno-Cruz, F.; Mendoza-Caamal, E.; Cicerón-Arellano, I.; Flores-Huacuja, M.; Baca, P.; et al. The genomic landscape of Mexican Indigenous populations brings insights into the peopling of the Americas. Nat. Commun. 2021, 12, 5942. [Google Scholar] [CrossRef]
- Popović, D.; Molak, M.; Ziółkowski, M.; Vranich, A.; Sobczyk, M.; Vidaurre, D.U.; Agresti, G.; Skrzypczak, M.; Ginalski, K.; Lamnidis, T.C.; et al. Ancient genomes reveal long-range influence of the pre-Columbian culture and site of Tiwanaku. Sci. Adv. 2021, 7, eabg7261. [Google Scholar] [CrossRef]
- Capodiferro, M.R.; Aram, B.; Raveane, A.; Migliore, N.R.; Colombo, G.; Ongaro, L.; Rivera, J.; Mendizábal, T.; Hernández-Mora, I.; Tribaldos, M.; et al. Archaeogenomic distinctiveness of the Isthmo-Colombian area. Cell 2021, 184, 1706–1723.e24. [Google Scholar] [CrossRef]
- Ribeiro-Dos-Santos, A.M.; Vidal, A.F.; Vinasco-Sandoval, T.; Guerreiro, J.; Santos, S.; de Souza, S.J.; Ribeiro-dos-Santos, Â. Exome Sequencing of Native Populations From the Amazon Reveals Patterns on the Peopling of South America. Front. Genet. 2020, 11, 548507. [Google Scholar] [CrossRef]
- Piaggi, P.; Köroğlu, Ç.; Nair, A.K.; Sutherland, J.; Muller, Y.L.; Kumar, P.; Hsueh, W.C.; Kobes, S.; Shuldiner, A.R.; Kim, H.I.; et al. Exome Sequencing Identifies a Nonsense Variant in DAO Associated with Reduced Energy Expenditure in American Indians. J. Clin. Endocrinol. Metab. 2020, 105, e3989–e4000. [Google Scholar] [CrossRef]
- Ioannidis, A.G.; Blanco-Portillo, J.; Sandoval, K.; Hagelberg, E.; Miquel-Poblete, J.F.; Moreno-Mayar, J.V.; Rodríguez-Rodríguez, J.E.; Quinto-Cortés, C.D.; Auckland, K.; Parks, T.; et al. Native American gene flow into Polynesia predating Easter Island settlement. Nature 2020, 583, 572–577. [Google Scholar] [CrossRef]
- Fernandes, D.M.; Sirak, K.A.; Ringbauer, H.; Sedig, J.; Rohland, N.; Cheronet, O.; Mah, M.; Mallick, S.; Olalde, I.; Culleton, B.J.; et al. A genetic history of the pre-contact Caribbean. Nature 2021, 590, 103–110. [Google Scholar] [CrossRef]
- Borda, V.; Alvim, I.; Mendes, M.; Silva-Carvalho, C.; Soares-Souza, G.B.; Leal, T.P.; Scliar, M.O.; Zamudio, R.; Zolini, C.; Padilla, C.; et al. The genetic structure and adaptation of Andean highlanders and Amazonians are influenced by the interplay between geography and culture. Proc. Natl. Acad. Sci. USA 2020, 117, 32557–32565. [Google Scholar] [CrossRef]
- Ávila-Arcos, M.C.; McManus, K.F.; Sandoval, K.; Rodríguez-Rodríguez, J.E.; Villa-Islas, V.; Martin, A.R.; Luisi, P.; Peñaloza-Espinosa, R.I.; Eng, C.; Huntsman, S.; et al. Population History and Gene Divergence in Native Mexicans Inferred from 76 Human Exomes. Mol. Biol. Evol. 2020, 37, 994–1006. [Google Scholar] [CrossRef]
- Zhou, S.; Xie, P.; Quoibion, A.; Ambalavanan, A.; Dionne-Laporte, A.; Spiegelman, D.; Bourassa, C.V.; Xiong, L.; Dion, P.A.; Rouleau, G.A. Genetic architecture and adaptations of Nunavik Inuit. Proc. Natl. Acad. Sci. USA 2019, 116, 16012–16017. [Google Scholar] [CrossRef]
- Vidal, E.A.; Moyano, T.C.; Bustos, B.I.; Pérez-Palma, E.; Moraga, C.; Riveras, E.; Montecinos, A.; Azócar, L.; Soto, D.C.; Vidal, M.; et al. Whole Genome Sequence, Variant Discovery and Annotation in Mapuche-Huilliche Native South Americans. Sci. Rep. 2019, 9, 2132. [Google Scholar] [CrossRef]
- Flegontov, P.; Altınışık, N.E.; Changmai, P.; Rohland, N.; Mallick, S.; Adamski, N.; Bolnick, D.A.; Broomandkhoshbacht, N.; Candilio, F.; Culleton, B.J.; et al. Palaeo-Eskimo genetic ancestry and the peopling of Chukotka and North America. Nature 2019, 570, 236–240. [Google Scholar] [CrossRef]
- Gnecchi-Ruscone, G.A.; Sarno, S.; De Fanti, S.; Gianvincenzo, L.; Giuliani, C.; Boattini, A.; Bortolini, E.; di Corcia, T.; Mellado, C.S.; Francia, T.J.D.; et al. Dissecting the Pre-Columbian Genomic Ancestry of Native Americans along the Andes–Amazonia Divide. Mol. Biol. Evol. 2019, 36, 1254–1269. [Google Scholar] [CrossRef]
- Reynolds, A.W.; Mata-Míguez, J.; Miró-Herrans, A.; Briggs-Cloud, M.; Sylestine, A.; Barajas-Olmos, F.; Garcia-Ortiz, H.; Rzhetskaya, M.; Orozco, L.; Raff, J.A.; et al. Comparing signals of natural selection between three Indigenous North American populations. Proc. Natl. Acad. Sci. USA 2019, 116, 9312–9317. [Google Scholar] [CrossRef]
- Barbieri, C.; Barquera, R.; Arias, L.; Sandoval, J.R.; Acosta, O.; Zurita, C.; Aguilar-Campos, A.; Tito-Álvarez, A.M.; Serrano-Osuna, R.; Gray, R.D.; et al. The Current Genomic Landscape of Western South America: Andes, Amazonia, and Pacific Coast. Mol. Biol. Evol. 2019, 36, 2698–2713. [Google Scholar] [CrossRef]
- Sánchez-Pozos, K.; Ortíz-López, M.G.; Peña-Espinoza, B.I.; de los Ángeles Granados-Silvestre, M.; Jiménez-Jacinto, V.; Verleyen, J.; Tekola-Ayele, F.; Sanchez-Flores, A.; Menjivar, M. Whole-exome sequencing in maya indigenous families: Variant in PPP1R3A is associated with type 2 diabetes. Mol. Genet. Genom. 2018, 293, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Mayar, J.V.; Potter, B.A.; Vinner, L.; Steinrücken, M.; Rasmussen, S.; Terhorst, J.; Kamm, J.; Albrechtsen, A.; Malaspinas, A.-S.; Sikora, M.; et al. Terminal Pleistocene Alaskan genome reveals first founding population of Native Americans. Nature 2018, 553, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.N.; Song, W.; Shetty, A.C.; Levano, K.S.; Cáceres, O.; Padilla, C.; Borda, V.; Tarazona, D.; Trujillo, O.; Sanchez, C.; et al. Evolutionary genomic dynamics of Peruvians before, during, and after the Inca Empire. Proc. Natl. Acad. Sci. USA 2018, 115, E6526–E6535. [Google Scholar] [CrossRef] [PubMed]
- Lindo, J.; Haas, R.; Hofman, C.; Apata, M.; Moraga, M.; Verdugo, R.A.; Watson, J.T.; Llave, C.V.; Witonsky, D.; Beall, C.; et al. The genetic prehistory of the Andean highlands 7000 years BP though European contact. Sci. Adv. 2018, 4, eaau4921. [Google Scholar] [CrossRef]
- Schroeder, H.; Sikora, M.; Gopalakrishnan, S.; Cassidy, L.M.; Maisano Delser, P.; Sandoval Velasco, M.; Schraiber, J.G.; Rasmussen, S.; Homburger, J.R.; Ávila-Arcos, M.C.; et al. Origins and genetic legacies of the Caribbean Taino. Proc. Natl. Acad. Sci. USA 2018, 115, 2341–2346. [Google Scholar] [CrossRef]
- de la Fuente, C.; Ávila-Arcos, M.C.; Galimany, J.; Carpenter, M.L.; Homburger, J.R.; Blanco, A.; Contreras, P.; Dávalos, D.C.; Reyes, O.; San Roman, M.; et al. Genomic insights into the origin and diversification of late maritime hunter-gatherers from the Chilean Patagonia. Proc. Natl. Acad. Sci. USA 2018, 115, E4006–E4012. [Google Scholar] [CrossRef]
- Moreno-Mayar, J.V.; Vinner, L.; de Barros Damgaard, P.; de la Fuente, C.; Chan, J.; Spence, J.P.; Allentoft, M.E.; Vimala, T.; Racimo, F.; Pinotti, T.; et al. Early human dispersals within the Americas. Science 2018, 362, eaav2621. [Google Scholar] [CrossRef]
- Posth, C.; Nakatsuka, N.; Lazaridis, I.; Skoglund, P.; Mallick, S.; Lamnidis, T.C.; Rohland, N.; Nägele, K.; Adamski, N.; Bertolini, E.; et al. Reconstructing the Deep Population History of Central and South America. Cell 2018, 175, 1185–1197.e22. [Google Scholar] [CrossRef]
- Peng, Q.; Schork, N.J.; Wilhelmsen, K.C.; Ehlers, C.L. Whole genome sequence association and ancestry-informed polygenic profile of EEG alpha in a Native American population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017, 174, 435–450. [Google Scholar] [CrossRef]
- Romero-Hidalgo, S.; Ochoa-Leyva, A.; Garcíarrubio, A.; Acuña-Alonzo, V.; Antúnez-Argüelles, E.; Balcazar-Quintero, M.; Barquera-Lozano, R.; Carnevale, A.; Cornejo-Granados, F.; Fernández-López, J.C.; et al. Demographic history and biologically relevant genetic variation of Native Mexicans inferred from whole-genome sequencing. Nat. Commun. 2017, 8, 1005. [Google Scholar] [CrossRef]
- Lindo, J.; Achilli, A.; Perego, U.A.; Archer, D.; Valdiosera, C.; Petzelt, B.; Mitchell, J.; Worl, R.; Dixon, E.J.; Fifield, T.E.; et al. Ancient individuals from the North American Northwest Coast reveal 10,000 years of regional genetic continuity. Proc. Natl. Acad. Sci. USA 2017, 114, 4093–4098. [Google Scholar] [CrossRef]
- Fehren-Schmitz, L.; Jarman, C.L.; Harkins, K.M.; Kayser, M.; Popp, B.N.; Skoglund, P. Genetic Ancestry of Rapanui before and after European Contact. Curr. Biol. 2017, 27, 3209–3215.e6. [Google Scholar] [CrossRef]
- Lindo, J.; Huerta-Sánchez, E.; Nakagome, S.; Rasmussen, M.; Petzelt, B.; Mitchell, J.; Cybulski, J.S.; Willerslev, E.; DeGiorgio, M.; Malhi, R.S. A time transect of exomes from a Native American population before and after European contact. Nat. Commun. 2016, 7, 13175. [Google Scholar] [CrossRef]
- Manousaki, D.; Kent, J.W., Jr.; Haack, K.; Zhou, S.; Xie, P.; Greenwood, C.M.; Brassard, P.; Newman, D.E.; Cole, S.; Umans, J.G.; et al. Toward Precision Medicine: TBC1D4 Disruption Is Common Among the Inuit and Leads to Underdiagnosis of Type 2 Diabetes. Diabetes Care 2016, 39, 1889–1895. [Google Scholar] [CrossRef]
- Dewey, F.E.; Murray, M.F.; Overton, J.D.; Habegger, L.; Leader, J.B.; Fetterolf, S.N.; O’Dushlaine, C.; van Hout, C.V.; Staples, J.; Gonzaga-Jauregui, C.; et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 2016, 354, aaf6814. [Google Scholar] [CrossRef]
- Mallick, S.; Li, H.; Lipson, M.; Mathieson, I.; Gymrek, M.; Racimo, F.; Zhao, M.; Chennagiri, N.; Nordenfelt, S.; Tandon, A.; et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 2016, 538, 201–206. [Google Scholar] [CrossRef]
- Zhou, S.; Xiong, L.; Xie, P.; Ambalavanan, A.; Bourassa, C.V.; Dionne-Laporte, A.; Spiegelman, D.; Gauthier, M.T.; Henrion, E.; Diallo, O.; et al. Increased Missense Mutation Burden of Fatty Acid Metabolism Related Genes in Nunavik Inuit Population. PLoS ONE 2015, 10, e0128255. [Google Scholar] [CrossRef]
- Rasmussen, M.; Sikora, M.; Albrechtsen, A.; Korneliussen, T.S.; Moreno-Mayar, J.V.; Poznik, G.D.; Zollikofer, C.P.E.; de León, M.S.P.; Allentoft, M.; Moltke, I.; et al. The ancestry and affiliations of Kennewick Man. Nature 2015, 523, 455–458. [Google Scholar] [CrossRef]
- Skoglund, P.; Mallick, S.; Bortolini, M.C.; Chennagiri, N.; Hünemeier, T.; Petzl-Erler, M.L.; Salzano, F.M.; Patterson, N.; Reich, D. Genetic evidence for two founding populations of the Americas. Nature 2015, 525, 104–108. [Google Scholar] [CrossRef]
- Malaspinas, A.S.; Lao, O.; Schroeder, H.; Rasmussen, M.; Raghavan, M.; Moltke, I.; Campos, P.; Sagredo, F.S.; Rasmussen, S.; Gonçalves, V.F.; et al. Two ancient human genomes reveal Polynesian ancestry among the indigenous Botocudos of Brazil. Curr. Biol. 2014, 24, R1035–R1037. [Google Scholar] [CrossRef]
- Raghavan, M.; DeGiorgio, M.; Albrechtsen, A.; Moltke, I.; Skoglund, P.; Korneliussen, T.S.; Grønnow, B.; Appelt, M.; Gulløv, H.C.; Friesen, T.M.; et al. The genetic prehistory of the New World Arctic. Science 2014, 345, 1255832. [Google Scholar] [CrossRef]
- Rasmussen, M.; Anzick, S.L.; Waters, M.R.; Skoglund, P.; DeGiorgio, M.; Stafford, T.W.; Rasmussen, S.; Moltke, I.; Albrechtsen, A.; Doyle, S.M.; et al. The genome of a Late Pleistocene human from a Clovis burial site in western Montana. Nature 2014, 506, 225–229. [Google Scholar] [CrossRef]
- Verdu, P.; Pemberton, T.J.; Laurent, R.; Kemp, B.M.; Gonzalez-Oliver, A.; Gorodezky, C.; Hughes, C.E.; Shattuck, M.R.; Petzelt, B.; Mitchell, J.; et al. Patterns of Admixture and Population Structure in Native Populations of Northwest North America. PLoS Genet. 2014, 10, e1004530. [Google Scholar] [CrossRef]
- Moreno-Estrada, A.; Gignoux, C.R.; Fernández-López, J.C.; Zakharia, F.; Sikora, M.; Contreras, A.V.; Acuña-Alonzo, V.; Sandoval, K.; Eng, C.; Romero-Hidalgo, S.; et al. The genetics of Mexico recapitulates Native American substructure and affects biomedical traits. Science 2014, 344, 1280–1285. [Google Scholar] [CrossRef]
- Moreno-Mayar, J.V.; Rasmussen, S.; Seguin-Orlando, A.; Rasmussen, M.; Liang, M.; Flåm, S.T.; Lie, B.A.; Gilfillan, G.D.; Nielsen, R.; Thorsby, E.; et al. Genome-wide Ancestry Patterns in Rapanui Suggest Pre-European Admixture with Native Americans. Curr. Biol. 2014, 24, 2518–2525. [Google Scholar] [CrossRef]
- Lazaridis, I.; Patterson, N.; Mittnik, A.; Renaud, G.; Mallick, S.; Kirsanow, K.; Sudmant, P.H.; Schraiber, J.G.; Castellano, S.; Lipson, M.; et al. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature 2014, 513, 409–413. [Google Scholar] [CrossRef]
- Ribeiro-dos-Santos, A.M.; de Souza, J.E.S.; Almeida, R.; Alencar, D.O.; Barbosa, M.S.; Gusmão, L.; Silva, W.A., Jr.; de Souza, S.J.; Silva, W.A.; Darnet, S.; et al. High-Throughput Sequencing of a South American Amerindian. PLoS ONE 2013, 8, e83340. [Google Scholar] [CrossRef]
- Moreno-Estrada, A.; Gravel, S.; Zakharia, F.; McCauley, J.L.; Byrnes, J.K.; Gignoux, C.R.; Ortiz-Tello, P.A.; Martínez, R.J.; Hedges, D.J.; Morris, R.W.; et al. Reconstructing the Population Genetic History of the Caribbean. PLoS Genet. 2013, 9, e1003925. [Google Scholar] [CrossRef]
- Reich, D.; Patterson, N.; Campbell, D.; Tandon, A.; Mazieres, S.; Ray, N.; Parra-Marín, M.V.; Rojas, W.; Duque, C.; Mesa, N.; et al. Reconstructing Native American population history. Nature 2012, 488, 370–374. [Google Scholar] [CrossRef]
- Patterson, N.; Moorjani, P.; Luo, Y.; Mallick, S.; Rohland, N.; Zhan, Y.; Genschoreck, T.; Webster, T.; Reich, D. Ancient Admixture in Human History. Genetics 2012, 192, 1065–1093. [Google Scholar] [CrossRef]
- Reich, D.; Green, R.E.; Kircher, M.; Krause, J.; Patterson, N.; Durand, E.Y.; Viola, B.; Briggs, A.W.; Stenzel, U.; Johnson, P.L.F.; et al. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature 2010, 468, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.; Li, Y.; Lindgreen, S.; Pedersen, J.S.; Albrechtsen, A.; Moltke, I.; Metspalu, M.; Metspalu, E.; Kivisild, T.; Gupta, R.; et al. Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature 2010, 463, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Silva-Zolezzi, I.; Hidalgo-Miranda, A.; Estrada-Gil, J.; Fernandez-Lopez, J.C.; Uribe-Figueroa, L.; Contreras, A.; Balam-Ortiz, E.; del Bosque-Plata, L.; Velazquez-Fernandez, D.; Lara, C.; et al. Analysis of genomic diversity in Mexican Mestizo populations to develop genomic medicine in Mexico. Proc. Natl. Acad. Sci. USA 2009, 106, 8611–8616. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.B.; Greene, A.C.; Prasad, D.V.; Jiang, X.; Greene, C.S. Responsible, practical genomic data sharing that accelerates research. Nat. Rev. Genet. 2020, 21, 615–629. [Google Scholar] [CrossRef]
- Wilson, S.L.; Way, G.P.; Bittremieux, W.; Armache, J.P.; Haendel, M.A.; Hoffman, M.M. Sharing biological data: Why, when, and how. FEBS Lett. 2021, 595, 847–863. [Google Scholar] [CrossRef]
- Crampton, P.; Parkin, C. Warrior genes and risk-taking science. N. Z. Med. J. 2007, 120, U2439. [Google Scholar]
- Hudson, M.; Garrison, N.A.; Sterling, R.; Caron, N.R.; Fox, K.; Yracheta, J.; Anderson, J.; Wilcox, P.; Arbour, L.; Brown, A.; et al. Rights, interests and expectations: Indigenous perspectives on unrestricted access to genomic data. Nat. Rev. Genet. 2020, 21, 377–384. [Google Scholar] [CrossRef]
- Alpaslan-Roodenberg, S.; Anthony, D.; Babiker, H.; Bánffy, E.; Booth, T.; Capone, P.; Deshpande-Mukherjee, A.; Eisenmann, S.; Fehren-Schmitz, L.; Frachetti, M.; et al. Ethics of DNA research on human remains: Five globally applicable guidelines. Nature 2021, 599, 41–46. [Google Scholar] [CrossRef]
- Séguin, B.; Hardy, B.J.; Singer, P.A.; Daar, A.S. Genomics, public health and developing countries: The case of the Mexican National Institute of Genomic Medicine (INMEGEN). Nat. Rev. Genet. 2008, 9, S5–S9. [Google Scholar] [CrossRef]
- Hetu, M.; Koutouki, K.; Joly, Y. Genomics for All: International Open Science Genomics Projects and Capacity Building in the Developing World. Front. Genet. 2019, 10, 95. [Google Scholar] [CrossRef]
- Séguin, B.; Hardy, B.J.; Singer, P.A.; Daar, A.S. Genomic medicine and developing countries: Creating a room of their own. Nat. Rev. Genet. 2008, 9, 487–493. [Google Scholar] [CrossRef]
- Helmy, M.; Awad, M.; Mosa, K.A. Limited resources of genome sequencing in developing countries: Challenges and solutions. Appl. Transl. Genom. 2016, 9, 15–19. [Google Scholar] [CrossRef]
- Meagher, K.M.; Lee, L.M. Integrating Public Health and Deliberative Public Bioethics: Lessons from the Human Genome Project Ethical, Legal, and Social Implications Program. Public Health Rep. 2016, 131, 44. [Google Scholar] [CrossRef]
- de Vries, J.; Bull, S.J.; Doumbo, O.; Ibrahim, M.; Mercereau-Puijalon, O.; Kwiatkowski, D.; Parker, M. Ethical issues in human genomics research in developing countries. BMC Med. Ethics 2011, 12, 5. [Google Scholar] [CrossRef]
- Mello, M.M.; Wolf, L.E. The Havasupai Indian Tribe Case—Lessons for Research Involving Stored Biologic Samples. N. Engl. J. Med. 2010, 363, 204–207. [Google Scholar] [CrossRef]
- Dalton, R. Tribe blasts “exploitation” of blood samples. Nature 2002, 420, 111. [Google Scholar] [CrossRef]
- Merriman, T.; Cameron, V. Risk-taking: Behind the warrior gene story. N. Z. Med. J. 2007, 120, U2440. [Google Scholar]
- The International Work Group for Indigenous Affairs (IWGIA). The Indigenous World 2021, 35th ed.; IWGIA: Copenhagen, Denmark, 2021; 824p, Available online: https://iwgia.org/doclink/iwgia-book-the-indigenous-world-2021-eng/eyJ0eXAiOiJKV1QiLCJhbGciOiJIUzI1NiJ9.eyJzdWIiOiJpd2dpYS1ib29rLXRoZS1pbmRpZ2Vub3VzLXdvcmxkLTIwMjEtZW5nIiwiaWF0IjoxNjI4ODM5NjM2LCJleHAiOjE2Mjg5MjYwMzZ9.z1CuM7PcT5CPkV0evx8ve88y6v0vmwDu_51JQ_lwAkM (accessed on 22 March 2022).
- Schauberger, P.; Walker, A.; Braglia, L.; Sturm, J.; Garbuszus, J.M.; Barbone, J.M. Openxlsx: Read, Write and Edit xlsx Files. 2021. Available online: https://CRAN.R-project.org/package=openxlsx (accessed on 11 March 2022).
- Wickham, H.; François, R.; Henry, L.; Müller, K.; RStudio. Dplyr: A Grammar of Data Manipulation. 2022. Available online: https://CRAN.R-project.org/package=dplyr (accessed on 11 March 2022).
- Wickham, H.; Chang, W.; Henry, L.; Pedersen, T.L.; Takahashi, K.; Wilke, C.; Woo, K.; Yutani, H.; Dunnington, D.; RStudio. Ggplot2: Create Elegant Data Visualisations Using the Grammar of Graphics. 2021. Available online: https://CRAN.R-project.org/package=ggplot2 (accessed on 11 March 2022).
- Slowikowski, K.; Schep, A.; Hughes, S.; Dang, T.K.; Lukauskas, S.; Irisson, J.-O.; Kamvar, Z.N.; Ryan, T.; Christophe, D.; Hiroaki, Y.; et al. Ggrepel: Automatically Position Non-Overlapping Text Labels with “ggplot2”. 2021. Available online: https://CRAN.Rproject.org/package=ggrepel (accessed on 11 March 2022).
- Wilkins, D.; Rudis, B. Treemapify: Draw Treemaps in “ggplot2”. 2021. Available online: https://CRAN.R-project.org/package=treemapify (accessed on 11 March 2022).
- Wilke, C.O. Cowplot: Streamlined Plot Theme and Plot Annotations for “ggplot2”. 2020. Available online: https://CRAN.Rproject.org/package=cowplot (accessed on 11 March 2022).
- Xiao, N.; Li, M. Ggsci: Scientific Journal and Sci-Fi Themed Color Palettes for “ggplot2”. 2018. Available online: https://CRAN.Rproject.org/package=ggsci (accessed on 11 March 2022).
- Wickham, H.; Girlich, M.; RStudio. Tidyr: Tidy Messy Data. 2022. Available online: https://CRAN.R-project.org/package=tidyr (accessed on 11 March 2022).
- Wickham, H.; Seidel, D.; RStudio. Scales: Scale Functions for Visualization. 2020. Available online: https://CRAN.R-project.org/package=scales (accessed on 11 March 2022).
- Xie, Y. Knitr: A General-Purpose Package for Dynamic Report Generation in R. 2021. Available online: https://yihui.org/knitr/ (accessed on 11 March 2022).
- Xie, Y. Dynamic Documents with R and Knitr, 2nd ed.; Chapman and Hall: London, UK; CRC: Boca Raton, FL, USA, 2015; Available online: https://www.routledge.com/Implementing-Reproducible-Research/Stodden-Leisch-Peng/p/book/9780367576172 (accessed on 11 March 2022).
- Stodden, V.; Leisch, F.; Peng, R.D. Implementing Reproducible Research; CRC Press: Boca Raton, FL, USA, 2014; 440p. [Google Scholar]
- South, A. Rnaturalearth: World Map Data from Natural Earth. 2017. Available online: https://CRAN.R-project.org/package=rnaturalearth (accessed on 11 March 2022).
- Pebesma, E. Simple Features for R: Standardized Support for Spatial Vector Data. R J. 2018, 10, 439–446. [Google Scholar] [CrossRef]
- Tennekes, M. tmap: Thematic Maps in R. J. Stat. Softw. 2018, 84, 1–39. [Google Scholar] [CrossRef]
- Bivand, R.; Rundel, C.; Pebesma, E.; Stuetz, R.; Hufthammer, K.O.; Giraudoux, P.; Davis, M.; Santilli, S. Rgeos: Interface to Geometry Engine—Open Source (‘GEOS’). 2021. Available online: https://CRAN.R-project.org/package=rgeos (accessed on 11 March 2022).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguilar-Ordoñez, I.; Guzmán-Linares, J.; Ballesteros-Villascán, J.; Mirón-Toruño, F.; Pérez-González, A.; García-López, J.; Cruz-López, F.; Morett, E. A Tale of Native American Whole-Genome Sequencing and Other Technologies. Diversity 2022, 14, 647. https://doi.org/10.3390/d14080647
Aguilar-Ordoñez I, Guzmán-Linares J, Ballesteros-Villascán J, Mirón-Toruño F, Pérez-González A, García-López J, Cruz-López F, Morett E. A Tale of Native American Whole-Genome Sequencing and Other Technologies. Diversity. 2022; 14(8):647. https://doi.org/10.3390/d14080647
Chicago/Turabian StyleAguilar-Ordoñez, Israel, Josué Guzmán-Linares, Judith Ballesteros-Villascán, Fernanda Mirón-Toruño, Alejandra Pérez-González, José García-López, Fabricio Cruz-López, and Enrique Morett. 2022. "A Tale of Native American Whole-Genome Sequencing and Other Technologies" Diversity 14, no. 8: 647. https://doi.org/10.3390/d14080647
APA StyleAguilar-Ordoñez, I., Guzmán-Linares, J., Ballesteros-Villascán, J., Mirón-Toruño, F., Pérez-González, A., García-López, J., Cruz-López, F., & Morett, E. (2022). A Tale of Native American Whole-Genome Sequencing and Other Technologies. Diversity, 14(8), 647. https://doi.org/10.3390/d14080647