Abstract
The Pleosporales is the most predominant order in the Dothideomycetes class, which contains over 4700 species that function in a variety of ways. The material used in this research was previously isolated from the Chinese white wax scale insect, and it was determined to be a Paraconiothyrium genus species that belonged to the Pleosporales order. For further molecular analysis, we assembled the complete mitochondrial genome of Paraconiothyrium sp. based on short reads of BGISEQ sequencing and subreads from Pacbio sequencing. The results showed that it was 42,734 bp in length and contained 8 open reading frames, 12 protein-coding genes and 31 non-coding genes. Phylogenetic analysis showed it was affiliated to the Pleosporales order and formed a sister relationship with Pithomyces chartarum. Compared to the seven other species in the Pleosporales order, Paraconiothyrium sp. has generally conserved gene content and structure, while the homologous blocks and gene order were shown to be significantly rearranged, in accordance with the species diversity in the Pleosporales order. In this study, we presented the first mitochondrial genome of Paraconiothyrium fungi to be reported, and we also showed gene order diversity in the Pleosporales order. These findings will lay the foundation for further species studies regarding molecular diversity and our understanding of species characteristics in the Paraconiothyrium genus.
1. Introduction
The Pleosporales order contains more than 4700 species, which account for nearly one-quarter of the Dothideomycetes class [1]. These species are present in various habitats, and they can exist as symbionts, parasites or endophytes of plant stems, plant leaves and insects [1,2]. As a member of the Pleosporales order, the Paraconiothyrium genus was found to be widely distributed, having been discovered in environments ranging from marine to soil. Additionally, instead of living an independent lifestyle, most species in this genus survive through reliance on a wide range of hosts, including plants, insects, etc. [3]. Commonly regarded as pathogens of plants, members of the Paraconiothyrium species can infect palm trees (Phoenix theophrasti), apple trees, pear trees and vines and cause spots or trunk damage [4,5]. These pathogens were found to affect humans with immunodeficiencies [6]. Moreover, they were found to exist inside insects, such as Acrida cinerea and Diaspidiotus sp. In insects, they were found to produce multiple chemical compounds, which were thought to be beneficial to the host insect without causing any disease [7,8]. Previously, we successfully isolated the fungi Paraconiothyrium sp. YMF1.07793 from the Chinese white wax scale insect (Ericerus pela) and revealed the existence of a symbiotic relationship between the fungus symbiont and the host based on amino acid and vitamin metabolic complementation [9].
The insect E. pela is well known as one of the most famous resource insects in China. It has great economic value due to the white wax it produces [10,11,12,13,14,15,16], which has been widely applied in Chinese medicine, the cosmetics industry and wax-printing products over the past thousand years. According to fossils of scale insects, the ancestors of scale insects were thought to dwell on the ground, and during this period, they came into contact with many microorganisms, especially when taking in nutrition from macrofungi or living in litter underground. Their transition from the ground to higher branches of plants with the prosperity of angiosperms thus led to a series of changes, including in their metabolism and physiology, and some microorganisms were subsequently retained as symbionts of insects. Paraconiothyrium sp. YMF1.07793 is considered to serve as a symbiont fungus in E. pela, although in most cases, Paraconiothyrium species are known to be pathogenic endophytes of plants [3].
Pleosporales species are exposed to diverse environments, and this environmental restriction consequently leads to diversification in the lifestyle and morphology of fungi in order for them to adapt. Therefore, an increasing number of unusual morphological characteristics of Pleosporales species are emerging [17]. For instance, species belonging to the Leptosphaeriaceae family are lightly pigmented and have multi-septate ascospores. Most Lophiostomataceae species usually contain ascomata that possess a compressed apex. Members of the Sporormiaceae family are heavily pigmented and are characterized by multi-septate ascospores which possess germ slits. Due to various inhabitation factors, diversified lifestyles and complicated morphological characteristics, some members of families such as the Venturiaceae family were miscategorized as belonging to the Pleosporales order [17,18]. At the same time, the formation of some morphological features of Pleosporales could be attributed to convergent evolution, such as fruiting body shapes [19]. Thus, it is difficult to identify the phylogenetic classification of these species using their morphological features. As has been established, the identification of Pleosporales species was originally based on their typical morphological characteristics, such as the presence of ascomata and hamathecium [20]. In this case, the diversity of the morphological characteristics in this order means they cannot be used for species identification and other corresponding studies. With the rapid development of sequencing technology that is currently taking place, genomic data could certainly provide information for species determination in a more precise way.
Compared with nuclear DNA, the mitochondrial genome has specific properties, such as smaller genome size, higher evolutionary rate and fewer gene recombination, which make it suitable for use in evolutionary biology research. With the advance in sequencing technology, fungal mitochondrial genomics has made great development in recent years and was applied to phylogenetic questions, including Pleosporales species [21]. Considering that Paraconiothyrium sp. is also a fungal species that belongs to the Pleosporales order, compared to its large and complicated nuclear genome, it is seemingly more suitable for use in analysis carried out with mitochondrial genomic data for the detection of its variety at the molecular level and phylogenetic relationships with other Pleosporales species. In this study, the whole mitochondrial genome of the fungi Paraconiothyrium sp. YMF1.07793 was assembled. Further analyses regarding species synteny and phylogeny were carried out for the determination of phylogenic positions and molecular diversity in Pleosporales species. This comparative analysis provided a more comprehensive perspective on the detection of species diversity within the Pleosporales order.
2. Methods
2.1. DNA Extraction and Sequencing
The fungus Paraconiothyrium sp. YMF1.07793 was isolated from E. pela in Yunnan Province (25°06′ N, 102°76′ E). It was deposited in the Microbial Library of the Germplasm Bank of Wild Species from Southwest China under the voucher number YMF1.07793. Genomic DNA of Paraconiothyrium sp. YMF1.07793 was extracted using CTAB lysis buffer [22].
The DNBSEQ library for the BGISEQ platform was constructed through high-density DNA nanochip technology. The SMRTbell library with 10–15 Kb fragments for the PacBio library was constructed using the SMRTbell Express Template Preparation Kit 2.0 (Pacific Biosciences, Menlo Park, CA, USA). After quality detection and assessment, the genomic DNA was sequenced based on both the BGISEQ and PacBio platforms.
2.2. Mitochondrial Genome Assembly
Here, 3907 Mb of raw data were obtained. The length of sequencing was 150 bp. The raw data from BGISEQ short reads were polished using the SOAPnuke software [23] to remove low-quality sequences and adaptors. Then, short reads were assembled into the mitochondrial genome using the SPAdes software (v3.11.0.) [24].
Then, all the subreads from PacBio sequencing were aligned with the mitochondrial genome assembled from BGISEQ short reads via local BLAST to obtain the mitochondrial subreads. These filtered subreads were assembled into the mitochondrial genome using Racon coupled with miniasm [25]. The mitochondrial genome was submitted to the GenBank database with the accession number OM617730.
2.3. Gene Annotation
Genome annotation was conducted using the MITOS web server and MFannot tool (v1.1). The annotation results were processed with additional artificial revision. The locations and lengths of essential mitochondrial genes were predicted using the MITOS WebServer (http://mitos2.bioinf.uni-leipzig.de/index.py, accessed on 1 March 2022). Open reading frames (ORFs) were detected using the ORF finder (https://www.ncbi.nlm.nih.gov/orffinder/, accessed on 1 March 2022). The complete mitochondrial Genome circular map was created on the webserver Organellar genome-draw (http://chlorobox.mpimp-golm.mpg.de/OGDraw.html, accessed on 1 March 2022).
2.4. Gene Component and Structure Analyses
The secondary structures of tRNA genes were depicted using the tRNAscan-SE search server (http://lowelab.ucsc.edu/tRNAscan-SE/index.html, accessed on 1 March 2022) with the default parameter settings. The tandem repeat units of the mitochondrial genome were analyzed using the Tandem Repeats Finder online server (http://tandem.bu.edu/trf/trf.html, accessed on 1 March 2022). The relative synonymous codon usage (RSCU) of Paraconiothyrium sp. was analyzed by importing all the sequences of mitochondrial protein-coding genes (PCGs) into the CodonW software (v1.4.2). The introns were identified from the annotation result of the MITOS web server and MFannot tool (v1.1). The analysis of the base composition skew was conducted manually according to the formula AT-skew = [A − T]/[A + T] and the formula GC-skew = [G − C]/[G + C] [26].
2.5. Phylogenetic Analysis
Phylogenetic analysis was carried out based on the 12 PCGs of the complete mitochondrial genome of Paraconiothyrium sp. and the 11 other species from the Pleosporales order (Table S1). The genome sequences were downloaded from the NCBI. Rhynchosporium orthosporum from the Ploettnerulaceae family was used as an outgroup. Multiple sequence alignment of the 12 homologous PCGs was performed using MUSCLE, and the redundant fragments were deleted manually [27]. A phylogenetic tree was constructed using Mega software (v.11) with the maximum likelihood method. The model applied was “cpREV + G + F”. The model with the lowest score for the Bayesian Information Criterion was selected.
2.6. Synteny Analysis
Based on phylogenetic analysis, we adopted the first eight Pleosporales species (which had a relatively close phylogenetic relationship with Paraconiothyrium sp.) in Table S1 to conduct synteny analysis. On the basis of the eight mitochondrial genomes downloaded from the NCBI, gene synteny analysis was carried out at the nucleic acid level using Mauve (v2.1.1).
2.7. Gene Rearrangement Analysis
Gene rearrangement analysis was conducted with the same eight Pleosporales species in synteny analysis. The essential mitochondrial genes (PCGs, tRNAs and rRNAs) were used for gene rearrangement analysis. Based on the eight mitochondrial genomes obtained from GenBank, the gene order of the selected species was indicated using colored fragments. The same types of genes were marked with the same color.
3. Results
3.1. General Genomic Features
The final complete mitochondrial genome assembled from PacBio subreads was 42,734 bp in length. There was one large sequence fragment with a length of 15,381 bp, which had the opposite direction to the mitochondrial genome assembled from short reads, indicating that there were differences between the mitochondrial genomes obtained from short reads and PacBio subreads.
The overall GC content was 27.86%, and the overall AT content was 72.14%. Additionally, the base composition was as follows: 35.77% A, 36.21% T, 14.14% C and 13.87% G (Table S2). The mitochondrial genome was composed of 8 open reading frames (ORFs) (orf100, orf112, orf113, orf125, orf133, orf144, orf215 and orf271), 12 protein-coding genes (PCGs) and 31 non-coding genes (Figure 1).
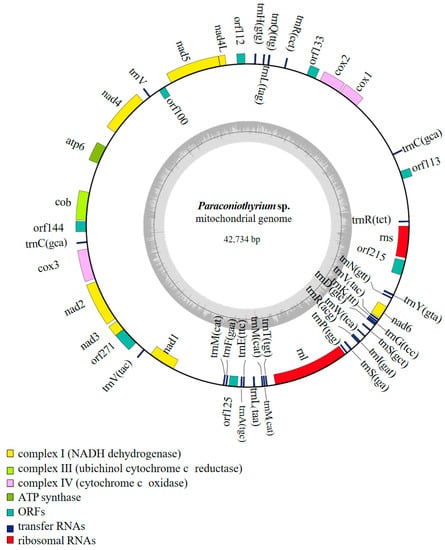
Figure 1.
Mitochondrial gene map of Paraconiothyrium sp. YMF1.07793.
The 12 PCGs included 7 NADH dehydrogenase genes (nad1, nad2, nad3, nad4, nad4L, nad5 and nad6), 3 cytochrome c oxidase genes (cox1, cox2 and cox3), 1 cytochrome c reductase gene (cob) and 1 ATP synthase synthesis gene (atp6). In addition to the 12 PCGs, 31 non-coding genes were identified, which included 2 ribosomal RNA genes (rrnL and rrnS) and 29 transfer RNA genes that transferred 18 amino acids (Table 1). The introns and intergenic regions were 23,747 bp in total, which accounted for 55.6% of the whole genome. There were four introns in the mitochondrial genome, and all the introns belonged to group I (GIY-YIG and LAGLIDADG).

Table 1.
Mitochondrial genome components of Paraconiothyrium sp.
3.2. Protein-Coding Genes and Codon Usage
The total length of the 12 PCGs was 12,139 bp, which accounted for 28.4% of the whole genome. The 12 PCGs had different sequence length sizes. The nad4L gene had the shortest sequence length of 270 bp, while the nad5 gene had the longest sequence length of 2183 bp. Except for introns within genes and the stop codons, the mitochondrial genome of Paraconiothyrium sp. encoded 3508 amino acids in total.
Within the 12 PCGs, there were three types of initiated codons. In total, 10 PCGs had the ORFs initiated with ATN codons (cox1, cox2, cox3, cob, nad2, nad4L, nad5 and nad6 with ATG, nad3 with ATA and nad4 with ATT), while the other 2 PCGs (nad1 and atp6) had the ORFs initiated with ACA and AAC, respectively.
According to the statistics of relative synonymous codon usage (RSCU), UUU of phenylalanine (Phe), UUA of Leucine (Leu) and UAU of tyrosine (Tyr) were the highest-frequency codons among all 12 PCGs. Leucine (Leu) accounted for the largest proportion of amino acid composition, 17.3%, which was followed by l-isoleucine (Ile) with 11.7% and tyrosine (Tyr) with 8.4%, while Tryptophan (Trp) accounted for the smallest proportion of 1.1% (Figure 2).
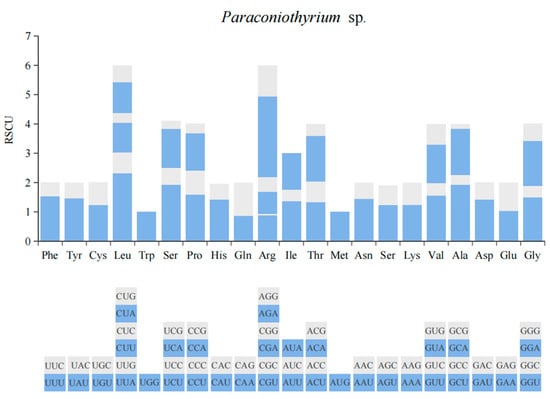
Figure 2.
Relative synonymous codon usage (RSCU).
3.3. rRNAs and tRNAs
The mitochondrial genome contained two ribosomal RNA genes, which included the large subunit ribosomal RNA gene (rrnL), which was located between trnT and trnP, and the small subunit ribosomal RNA gene (rrnS), which was located between orf215 and trnR. The length of rrnL was 3127 bp, while the length of rrnS was 1366 bp. The A + T contents of rrnL and rrnS were 65.94% and 65.3%, respectively.
The mitochondrial genome of Paraconiothyrium sp. contained 29 tRNA genes which ranged from 60 bp (trnR) to 85 bp (trnS and trnY). According to the analysis result from tRNAscan-SE, 20 standard aa were decoded by the remaining 28 tRNAs (including 1 possible suppressor tRNA), except for 1 tRNA with an unknown isotype. The tRNAs that conveyed trnL, trnS and trnY possessed irregular secondary structures. Additionally, the other 21 tRNAs presented typical cloverleaf secondary structures, comprising one amino-acyl (AA) arm, one TΨC (T) arm, one anticodon (AC) arm, one dihydorouridine (DHU) arm and one variable (V) loop (Figure S1).
3.4. Phylogenetic Analysis
According to the phylogenetic analysis, the 12 species belonging to the Pleosporales order, including Paraconiothyrium sp. YMF1.07793, were clustered into the main clade of the phylogenetic tree, and Rhynchosporium orthosporum, which belongs to the Ploettnerulaceae family, was separated as the outgroup. Paraconiothyrium sp. YMF1.07793 and Pithomyces chartarum formed a sister group with a 100% bootstrap value. In contrast, species belonging to the Pleosporaceae family in the Pleosporales order (Bipolaris sorokiniana, Bipolaris oryzae, Bipolaris cookei, Stemphylium lycopersici and Alternaria alternata) were shown to have relatively distant phylogenetic relationships with Paraconiothyrium sp. YMF1.07793 (Figure 3).
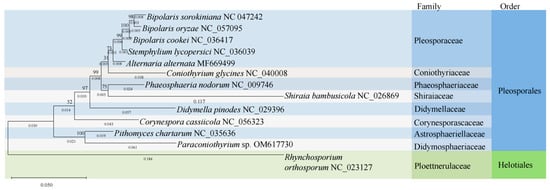
Figure 3.
Phylogenetic tree inferred from nucleotide sequences of 12 PCGs using the ML method. Bootstrap support values are indicated on branches.
3.5. Synteny Analysis
The synteny analysis of species from eight families in the Pleosporales order showed that the total number of homologous blocks in each mitochondrial genome varied from 13 (Coniothyrium glycines, Corynespora cassiicola, Phaeosphaeria nodorum, Shiraia bambusicola) to 16 (Pithomyces chartarum). Four homologous blocks existed in all eight species. The relative positions and sizes of these homologous blocks were dramatically different in each species. In particular, the last block in Stemphylium lycopersici, colored purple in the figure, was the largest homologous block (Figure 4).
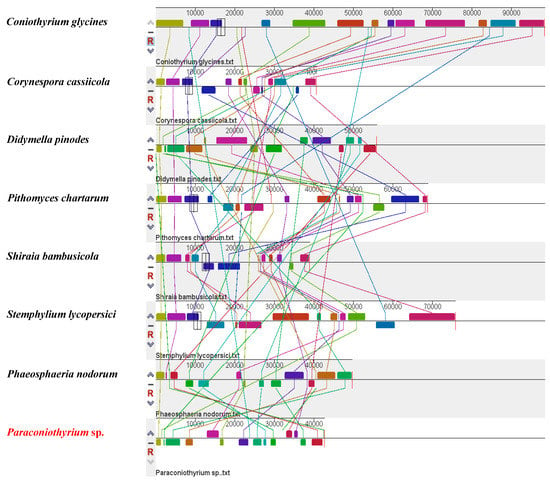
Figure 4.
Whole mitochondrial alignments of 8 Pleosporales species.
3.6. Gene Rearrangement
According to gene arrangement analysis, the gene order of tRNAs from rrnL to the first PCG was generally conserved among the seven species, except for Paraconiothyrium sp., in which nad1 was inserted within 10 tRNAs. The gene order from the last PCG nad6 to trnP also exhibited similar conservation among all eight species (Figure 5). It was found that gene rearrangement frequently occurred among PCGs (mostly cox genes and nad genes) in the mitochondrial genomes. Additionally, this rearrangement took place in a variety of patterns. By comparing Paraconiothyrium sp. with its sister group, Pithomyces chartarum, it could be found that one of the most obvious differences was between nad4L and cox2, in which four tRNA genes were inserted in Paraconiothyrium sp., and this also led to a total increase in gene number. However, Pithomyces chartarum was mainly composed of a succession of PCGs. The other obvious gene order difference was shown in Paraconiothyrium sp., in which a series of genes (cox3-trnC-cob-atp6) was inserted between nad2 and nad4, while Pithomyces chartarum was composed of a series of nad genes.
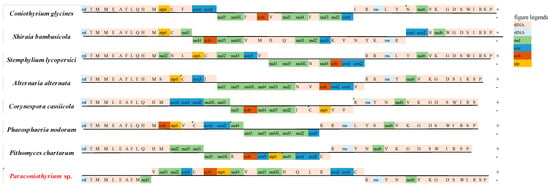
Figure 5.
Mitochondrial gene order rearrangements among 8 Pleosporales species. The genes on the H-strand (+) were shown above the line, and the genes on the L-strand (−) were shown below the line.
4. Discussion
In this study, we assembled the complete mitochondrial genome of Paraconiothyrium sp. YMF1.07793 isolated from E. pela based on subreads from the PacBio sequence. This is the first completely annotated mitochondrial genome of fungi from the Didymosphaeriacea family in the Pleosporales order. It provides a valid fungi mitochondrial genome that will contribute to further species diversity studies regarding both parasitism and symbiosis. In terms of its basic mitochondrial genome characteristics, Paraconiothyrium sp. YMF1.07793 is similar to other species in the Pleosporales order, as it exhibits normal mitochondrial gene content and a moderately sized mitochondrial genome among the Pleosporales order.
In the Pleosporales order, the mitochondrial genome size varies greatly, from the smallest mitochondrial genome size of Pyrenophora tritici-repentis being 16,417 bp to the largest one of Pyrenophora teres f. teres being 221,815 bp. These species were shown to exhibit similar gene content but dramatically different mitochondrial genome sizes, which means there is no explicit correlation between mitochondrial genome size and gene content. According to the statistics of mitochondrial genome component of Pyrenophora teres f. teres (221,815 bp in length), Stemphylium lycopersici (75,911 bp in length) and Paraconiothyrium sp. (42,734 bp in length), the total length of essential genes (PCGs, tRNAs and rRNAs) were 40,747 bp and 23,772 bp, and 18,987 bp separately. Apparently, these essential genes did not respond to the variation of mitochondrial genome size. Additionally, from the statistics of some Pleosporales species, we found that species with larger mitochondrial genome sizes always had more introns (Table S3). This evidence showed that differences in mitochondrial genome length could be related to the variation in intergenic regions and introns [28,29,30]. Moreover, two important Pleosporales species, Leptosphaerulina chartarum and Curvularia trifolii, isolated from tobacco, could serve as symbiont endophytic fungi to assist in the growth of tobacco and help tobacco tissue to resist biotic or abiotic stress [31]. They were originally considered to cause global crop diseases. According to a previous study, the mitochondrial genomes of the two species were both of moderate size, being 68,926 bp and 59,100 bp, respectively. As symbionts, these two endophytic fungi contain complex metabolic regulation mechanisms and should contain a large number of regulatory mechanisms that reflect relatively large mitochondrial genome sizes. However, they have medium-sized mitochondrial genomes, meaning there is no obvious relation between the mitochondrial genome size and a fungus’s lifestyle, no matter whether it is an independent organism in an external environment or whether it lives as a parasite in vivo in hosts. This could also prove the view mentioned above: that intergenic regions, as well as introns, influence the size of a mitochondrial genome. Considering the fact that some species could be endophytic fungi that lack some genes and functions which could be supplemented by the host, this lack does have a possibility of resulting in variations in mitochondrial genomes (including variations in genome size).
In the mitochondrial genome of Paraconiothyrium sp., the codons of UUU, UUA and UAU were in the highest frequency (Figure 2), as these codons are completely comprised of A and U (T), it could reveal the high A + T content of PCGs. According to the statistics of base composition, not just PCGs but the complete mitochondrial genome had high A + T content. Additionally, the high A + T content of the mitochondrial genome could be a general phenomenon of most fungal species [32]. Additionally, other studies reported that this high A + T content could represent an adaptation to nitrogen deficiency, which was in accordance with the fact that the host insect, which contained endogenous fungi inside, was always in lack of nitrogen [33]. Moreover, mutations are more likely to take place in sequences with high A + T content, since the A − T base pair (which contains two hydrogen bonds) is less stable than the G − C base pair (which contains three hydrogen bonds) [34].
In terms of the detailed mitochondrial genome characteristics of Paraconiothyrium sp. YMF1.07793, it contained 12 functional protein-coding genes (3 cytochrome c oxidase genes, 7 NADH dehydrogenase genes, an ATPase 6 gene and a cytochrome b gene) and 31 non-coding genes (29 tRNAs and 2 rRNAs), which were essentially identical to the mitochondrial genomes in other Pleosporales-order species. According to the annotation results, the atp8 gene was missing, and there was only an atp6 gene in Paraconiothyrium sp.; we did not find an atp8 gene in any of the other Pleosporales-order species. Therefore, we believed that there was no atp8 gene present in Paraconiothyrium sp. However, some species in the Dothideomycetes class, such as Zymoseptoria tritici and Zasmidium cellare, could contain an ATPase 8 gene and an ATPase 9 gene, which could possibly indicate that these species require a more complex energy metabolism and that they face multiple types of environmental stress [35]. Considering that we isolated Paraconiothyrium sp. from the scale insect E. pela and it was found that the two species had a symbiotic relationship, we determined that the deficiency regarding energy and nutrition in the fungi could possibly be supplemented by its insect host, and this could explain the lack of an atp8 gene in Paraconiothyrium sp.
In addition to the general conservation in gene content among Pleosporales-order species, based on an analysis regarding synteny and gene rearrangement, Paraconiothyrium sp. did undergo significant rearrangement regarding homologous blocks and gene orders, which implied a significant amount of gene rearrangement occurred. However, the rearrangement of gene order and homologous blocks occurred to totally different extents: homologous blocks were greatly rearranged, which was much more obvious than the extent to which changes to gene orders occurred. Although synteny analysis had adopted the whole mitochondrial genome (including mitochondrial genes and intergenic regions) into the analysis, the analysis of gene order rearrangement only took essential genes (PCGs, tRNAs and rRNAs) into consideration. It seemed like intergenic regions could be the reason for such great variations among species, but we did not find obvious evidence supporting this. According to the analysis result, only four homologous blocks were exhibited in all eight species. However, it could be found that there did exist a series of homologous blocks in several species that showed conservative gene composition of Pleosporales order. Generally, the tRNAs in the mitochondrial genomes of Pleosporales species were classified into two groups: the large clusters of tRNAs located beside the PCGs, and a few tRNAs inserted into PCGs. The two groups were also found in the Eurotiomycetes class and Sordariomycetes class [22]. Significantly, except for Coniothyrium glucines [36], the gene nad1 in all the species we adopted was located at the border of the gene cluster, and the genes around were distributed in two complement strains. Additionally, in many cases, trnV was located similarly at the border as well (Figure 5), which made these genes (including the repeat sequences around) potential recombinational hotspots for Pleosporales species because the gene itself as well as repeat sequences could contribute to gene shuffling (in Paraconiothyrium sp. there were repeat sequences in the region of nad1-trnV and the repeat sequences were detected by the website https://www.novoprolabs.com/tools/repeats-sequences-finder, accessed on 22 April 2022) [37,38,39]. As more and more unprecedented Pleosporales species and new lineages are continuously emerging, analyses regarding gene rearrangement and homologous blocks are still worthy of detecting to find out recombinational hotspots and rearrangement patterns a step further.
However, it is worth noting that the mitochondrial genome of Paraconiothyrium sp. YMF1.07793 was assembled based on PacBio sequences, and it showed a sequence fragment in the opposite direction to that assembled from BGISEQ sequences. We found this fragment had no influence on gene orders.
Species belonging to the Pleosporales order are widely distributed and live in a variety of habitats, from living in natural environments, e.g., marine [40,41,42,43], to existing as endosymbionts inside host insects, and Paraconiothyrium sp. has been proved to serve as an essential nutrition provider of E. pela according to our previous study [10]. Regardless of whether the responses related to fungi’s metabolic pathways are caused by environments or by hosts, their responses would lead to a requirement of energy, which is generally controlled by mitochondrion. Perhaps this also implies the genetic variation, including mitochondrial gene variation to some extent, and could finally result in species diversity.
Interestingly, compared to the Pleosporales-order species whose rearrangement has always been related to PCGs, in some other species, gene rearrangement related to other regions, such as non-coding genes, takes place. For instance, Saissetia coffeae, another scale insect belonging to the Coccoidea family, has been confirmed to have fragments filled with tRNAs that are involved in gene rearrangement [33]. It implied that the mitochondrion of hosts and endogenous fungi were under different regulation mechanisms.
In terms of high-level classification, despite the fact that analysis based on mitochondrial DNA sequences has been proved to be an essential tool in solving the remaining taxonomy issues, considering mutual regulation and cooperation that occurs between nuclear genomes and mitochondrial genomes, nuclear genomic data still need to be taken into consideration in the future. Equipped with multiple genomic data and typical morphological knowledge, more comprehensive and precise detection concerning species evolution and diversity in the Pleosporales order could take place.
Supplementary Materials
The following supporting material can be downloaded at: https://www.mdpi.com/article/10.3390/d14080601/s1. Figure S1. The tRNAs’ secondary structures of Paraconiothyrium sp. predicted using MITOS2, and the amino acid of each tRNA carried could be seen in the upside of the corresponding structure. Dashes between bases represent Watson–Crick bonds. Table S1. Phylogenetic analysis species. Table S2. Base composition and skew. Table S3. The representative Pleosporales species’ mitochondrial genome size and the number of introns.
Author Contributions
Conceptualization, P.Y.; Methodology, P.Y. and C.F.; Writing—Original Draft Preparation, J.A.; Writing—Review and Editing, P.Y., J.A. and C.F.; Data Curation, J.A., Z.F. and H.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Applied Basic Research Foundation of Yunnan Province [grant number 202101AS070042], the Young- and Middle-Aged Academic and Technical Leaders Reserve Talent Project of Yunnan Province [grant number 202105AC160031] and the National Natural Science Foundation of China [grant number 31572337].
Institutional Review Board Statement
Ethical review and approval were waived for this study, as E. pela is not an endangered species and it is cultured by humans. In addition, it is a serious pest in the field of landscaping and is also an invasive pest in some cities. No specific permits were required for this study.
Data Availability Statement
The data presented in this study are available in the NCBI GenBank (accession number: OM617730).
Acknowledgments
We thank Yang Lv of Shanghai Fengyuan Biotechnology LTD for providing help in the assembly of the mitochondrial genome.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhang, Y.; Schoch, C.; Fournier, J.; Crous, P.; de Gruyter, J.; Woudenberg, J.; Hirayama, K.; Tanaka, K.; Pointing, S.; Spatafora, J.; et al. Multi-locus phylogeny of Pleosporales: A taxonomic, ecological and evolutionary re-evaluation. Stud. Mycol. 2009, 64, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Câmara, M.P.; Palm, M.E.; van Berkum, P.; O’Neill, N.R. Molecular phylogeny of Leptosphaeria and Phaeosphaeria. Mycologia 2002, 94, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shao, S.; Liu, C.; Song, Z.; Liu, S.; Wu, S. The genus Paraconiothyrium: Species concepts, biological functions, and secondary metabolites. Crit. Rev. Microbiol. 2021, 47, 781–810. [Google Scholar] [CrossRef] [PubMed]
- Cloete, M.; Fourie, P.H.; Damm, U.; CROUS, P.W.; MOSTERT, L. Fungi associated with die-back symptoms of apple and pear trees, a possible inoculum source of grapevine trunk disease pathogens. Phytopathol. Mediterr. 2011, 50, S176–S190. [Google Scholar]
- Ligoxigakis, E.K.; Papaioannou, I.A.; Markakis, E.A.; Typas, M.A. First report of leaf spot of Phoenix theophrasti Caused by Paraconiothyrium variabile in Greece. Plant Dis. 2013, 97, 1250. [Google Scholar] [CrossRef]
- Gordon, R.A.; Sutton, D.A.; Thompson, E.H.; Shrikanth, V.; Verkley, G.J.M.; Stielow, J.B.; Mays, R.; Oleske, D.; Morrison, L.K.; Lapolla, W.J.; et al. Cutaneous phaeohyphomycosis caused by Paraconiothyrium cyclothyrioides. J. Clin. Microbiol. 2012, 50, 3795–3798. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-X.; Wang, L.; Chen, J.-F.; Guo, Z.-Y.; Tu, X.; Deng, Z.-S.; Zou, K. Paraconfuranones A-H, eight new furanone analogs from the insect-associated fungus Paraconiothyrium brasiliense MZ-1. Magn. Reason. Chem. 2015, 53, 317–322. [Google Scholar] [CrossRef]
- Ren, F.; Chen, S.; Zhang, Y.; Zhu, S.; Xiao, J.; Liu, X.; Su, R.; Che, Y. Hawaiienols A-D, highly oxygenated p-terphenyls from an insect-associated fungus, Paraconiothyrium hawaiiense. J. Nat. Prod. 2018, 81, 1752–1759. [Google Scholar] [CrossRef]
- Fu, Z.Y.; An, J.Q.; Liu, W.; Zhang, H.P.; Yang, P. Genomic Analyses of the Fungus Paraconiothyrium sp. Isolated from the Chinese White Wax Scale Insect Reveals Its Symbiotic Character. Genes 2022, 13, 338. [Google Scholar] [CrossRef]
- Yang, P.; Yu, S.; Hao, J.; Liu, W.; Zhao, Z.; Zhu, Z.; Sun, T.; Wang, X.; Song, Q. Genome sequence of the Chinese white wax scale insect Ericerus pela: The first draft genome for the Coccidae family of scale insects. GigaScience 2019, 8, 8. [Google Scholar] [CrossRef]
- Yang, P.; Zhu, J.-Y.; Gong, Z.-J.; Xu, D.-L.; Chen, X.-M.; Liu, W.-W.; Lin, X.-D.; Li, Y.-F. Transcriptome analysis of the Chinese white wax scale Ericerus pela with focus on genes involved in wax biosynthesis. PLoS ONE 2012, 7, e35719. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sun, T.; Wang, X.Q.; Zhao, Z.L.; Yu, S.H.; Yang, P.; Chen, X.M. A Lethal Fungus Infects the Chinese White Wax Scale Insect and Causes Dramatic Changes in the Host Microbiota. Sci. Rep. 2018, 8, 5324. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Chen, X.M. Protein profiles of Chinese white wax scale, Ericerus pela, at the male pupal stage by high-throughput proteomics. Arch. Insect Biochem. Physiol. 2014, 87, 214–233. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-H.; Yang, P.; Sun, T.; Qi, Q.; Wang, X.-Q.; Xu, D.-L.; Chen, X.-M. Identification and evaluation of reference genes in the Chinese white wax scale insect Ericerus pela. Springerplus 2016, 5, 791. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.A.; Okusu, A.; Normark, B.B. Molecular phylogenetics of Aspidiotini armored scale insects (Hemiptera: Diaspididae) reveals rampant paraphyly, curious species radiations, and multiple origins of association with Melissotarsus ants (Hymenoptera: Formicidae). Mol. Phylogenet. Evol. 2018, 129, 291–303. [Google Scholar] [CrossRef]
- Yang, P.; Chen, X.M.; Liu, W.W.; Feng, Y.; Sun, T. Transcriptome analysis of sexually dimorphic Chinese white wax scale insects reveals key differences in developmental programs and transcription factor expression. Sci. Rep. 2015, 5, 8141. [Google Scholar] [CrossRef]
- Zhang, Y.; Crous, P.W.; Schoch, C.L.; Hyde, K.D. Pleosporales. Fungal Divers. 2012, 53, 1–221. [Google Scholar] [CrossRef]
- Kruys, A.; Eriksson, O.E.; Wedin, M. Phylogenetic relationships of coprophilous Pleosporales (Dothideomycetes, Ascomycota), and the classification of some bitunicate taxa of unknown position. Mycol. Res. 2006, 110, 527–536. [Google Scholar] [CrossRef]
- Hawksworth, D.L.; Lagreca, S. New bottles for old wine: Fruit body types, phylogeny, and classification. Mycol. Res. 2007, 111, 999–1000. [Google Scholar] [CrossRef]
- Liew, E.C.; Aptroot, A.; Hyde, K.D. Phylogenetic significance of the pseudoparaphyses in Loculoascomycete taxonomy. Mol. Phylogenetics Evol. 2000, 16, 392–402. [Google Scholar] [CrossRef]
- Shen, X.Y.; Li, T.; Chen, S.; Fan, L.; Gao, J.; Hou, C.L. Characterization and phylogenetic analysis of the mitochondrial genome of Shiraia bambusicola reveals special features in the order of pleosporales. PLoS ONE 2015, 10, e0116466. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yu, S.-H.; Zhang, H.-P.; Fu, Z.-Y.; An, J.-Q.; Zhang, J.-Y.; Yang, P. Two Cladosporium Fungi with Opposite Functions to the Chinese White Wax Scale Insect Have Different Genome Characters. J. Fungi 2022, 8, 286. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018, 7, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome. Res. 2017, 27, 737–746. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Litter, J.; Keszthelyi, A.; Hamari, Z.; Pfeiffer, I.; Kucsera, J. Differences in mitochondrial genome organization of Cryptococcus neoformans strains. Antonie Van Leeuwenhoek 2005, 88, 249–255. [Google Scholar] [CrossRef]
- Wang, Y.; Zeng, F.; Hon, C.C.; Zhang, Y.; Leung, F.C. The mitochondrial genome of the Basidiomycete fungus Pleurotus ostreatus (oyster mushroom). FEMS Microbiol. Lett. 2008, 280, 34–41. [Google Scholar] [CrossRef][Green Version]
- Formighieri, E.F.; Tiburcio, R.A.; Armas, E.D.; Medrano, F.J.; Shimo, H.; Carels, N.; Góes-Neto, A.; Cotomacci, C.; Carazzolle, M.F.; Sardinha-Pinto, N.; et al. The mitochondrial genome of the phytopathogenic basidiomycete Moniliophthora perniciosa is 109 kb in size and contains a stable integrated plasmid. Mycol. Res. 2008, 112, 1136–1152. [Google Scholar] [CrossRef]
- Yuan, X.-L.; Cao, M.; Shen, G.-M.; Zhang, H.-B.; Du, Y.-M.; Zhang, Z.-F.; Li, Q.; Gao, J.-M.; Xue, L.; Wang, Z.-P.; et al. Characterization of Nuclear and Mitochondrial Genomes of Two Tobacco Endophytic Fungi Leptosphaerulina chartarum and Curvularia trifolii and Their Contributions to Phylogenetic Implications in the Pleosporales. Int. J. Mol. Sci. 2020, 21, 2461. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Kong, W.S.; Sung, G.H. Complete mitochondrial genome of the entomopathogenic fungus Beauveria pseudobassiana (Ascomycota, Cordycipitaceae). Mitochondrial DNA. 2015, 26, 777–778. [Google Scholar] [CrossRef]
- Lu, C.; Huang, X.; Deng, J. The challenge of Coccidae (Hemiptera: Coccoidea) mitochondrial genomes: The case of Saissetia coffeae with novel truncated tRNAs and gene rearrangements. Int. J. Biol. Macromol. 2020, 158, 854–864. [Google Scholar] [CrossRef]
- Mohajeri, A.; Nobandegani, F.F. Detection and evaluation of hydrogen bond strength in nucleic acid base pairs. J. Phys. Chem. A 2008, 112, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.B.; McCorison, C.B.; Cavaletto, J.R.; Culley, D.E.; LaButti, K.; Baker, S.; Grigoriev, I. The mitochondrial genome of the ethanol-metabolizing, wine cellar mold Zasmidium cellare is the smallest for a filamentous ascomycete. Fungal Biol. 2016, 120, 961–974. [Google Scholar] [CrossRef]
- Stone, C.L.; Frederick, R.D.; Tooley, P.W.; Luster, D.G.; Campos, B.; Winegar, R.A.; Melcher, U.; Fletcher, J.; Blagden, T. Annotation and analysis of the mitochondrial genome of Coniothyrium glycines, causal agent of red leaf blotch of soybean, reveals an abundance of homing endonucleases. PLoS ONE 2018, 13, e0207062. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Wu, Y.; Yang, C.; Gu, X.; Wilson, J.J.; Li, H.; Cai, W.; Yang, H.; Song, F. Evolution of tRNA gene rearrangement in the mitochondrial genome of ichneumonoid wasps (Hymenoptera: Ichneumonoidea). Int. J. Biol. Macromol. 2020, 164, 540–547. [Google Scholar] [CrossRef]
- Gong, L.; Shi, W.; Si, L.Z.; Kong, X.Y. Rearrangement of Mitochondrial Genome in Fishes. Dongwuxue Yanjiu 2013, 34, 666–673. [Google Scholar] [CrossRef]
- Lee, Y.P.; Kim, S.; Lim, H.; Ahn, Y.; Sung, S.K. Identification of mitochondrial genome rearrangements unique to novel cytoplasmic male sterility in radish (Raphanus sativus L.). Theor. Appl. Genet. 2009, 118, 719–728. [Google Scholar] [CrossRef]
- Amend, A.; Burgaud, G.; Cunliffe, M.; Edgcomb, V.P.; Ettinger, C.L.; Gutiérrez, M.H.; Heitman, J.; Hom, E.F.Y.; Ianiri, G.; Jones, A.C.; et al. Fungi in the Marine Environment: Open Questions and Unsolved Problems. mBio 2019, 10, e01189-18. [Google Scholar] [CrossRef]
- Cao, F.; Meng, Z.H.; Wang, P.; Luo, D.Q.; Zhu, H.J. Dipleosporalones A and B, Dimeric Azaphilones from a Marine-Derived Pleosporales sp. Fungus. J. Nat. Prod. 2020, 83, 1283–1287. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, H.; Ye, J.; Wu, X.; Wang, W.; Lin, H.; Yan, X.; Lazaro, J.; Wang, T.; Naman, C.; et al. Cytotoxic Polyketide Metabolites from a Marine Mesophotic Zone Chalinidae Sponge-Associated Fungus Pleosporales sp. NBUF144. Mar. Drugs 2021, 19, 186. [Google Scholar] [CrossRef] [PubMed]
- Prachyawarakorn, V.; Mahidol, C.; Sureram, S.; Sangpetsiripan, S.; Wiyakrutta, S.; Ruchirawat, S.; Kittakoop, P. Diketopiperazines and phthalides from a marine derived fungus of the order pleosporales. Planta Med. 2008, 74, 69–72. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).