
The Chromosome-Level Genome of Elaeagnus moorcroftii Wall., an Economically and Ecologically Important Tree Species in Drylands


 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Sequencing
2.2. Genome Assembly and Assessment
2.3. Genome Annotation
2.4. Comparative and Evolutionary Genomic Analysis
2.5. Whole Genome Duplication Analysis
3. Results
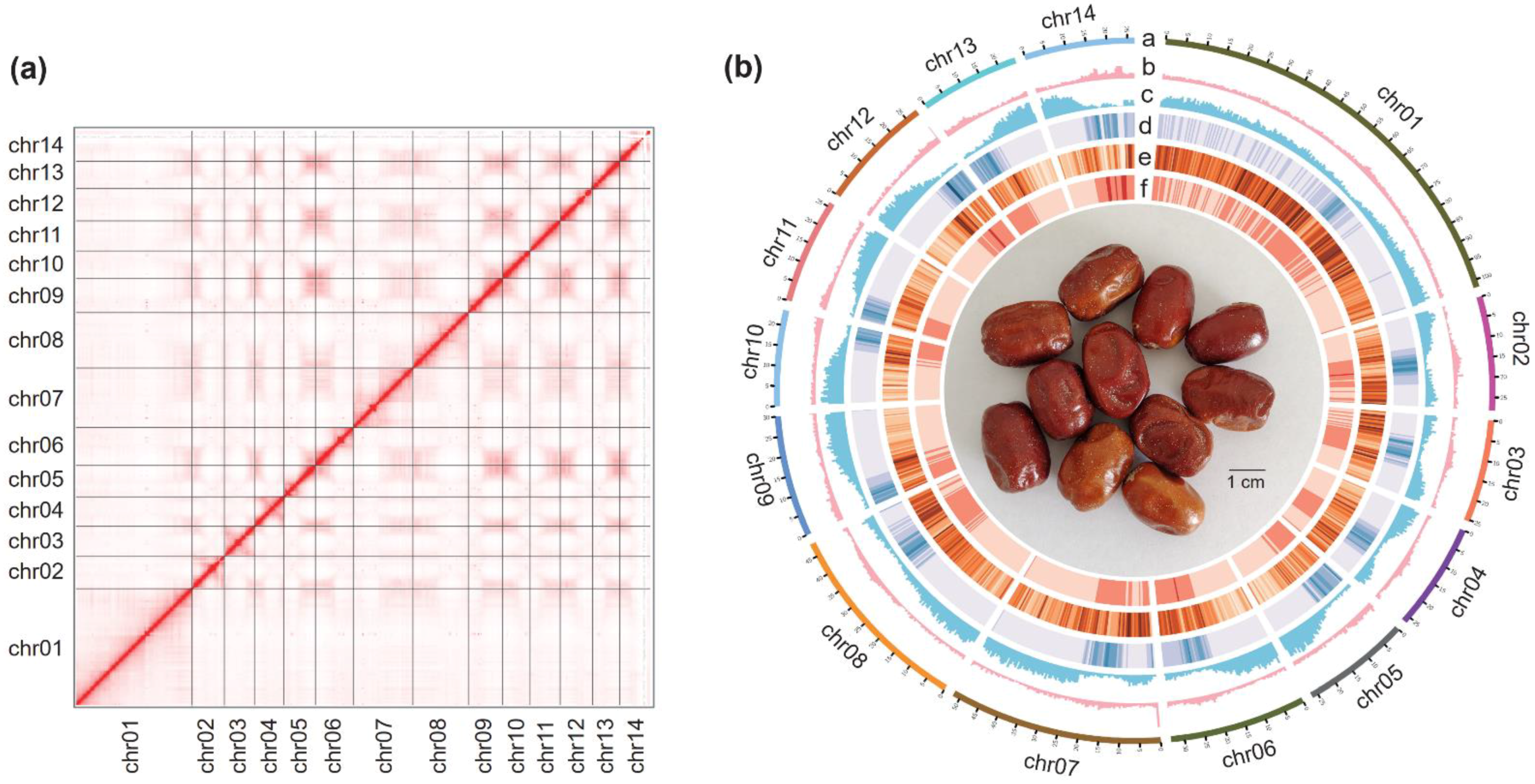
3.1. Genome Assembly of E. Moorcroftii
3.2. Annotation of the E. Moorcroftii Genome
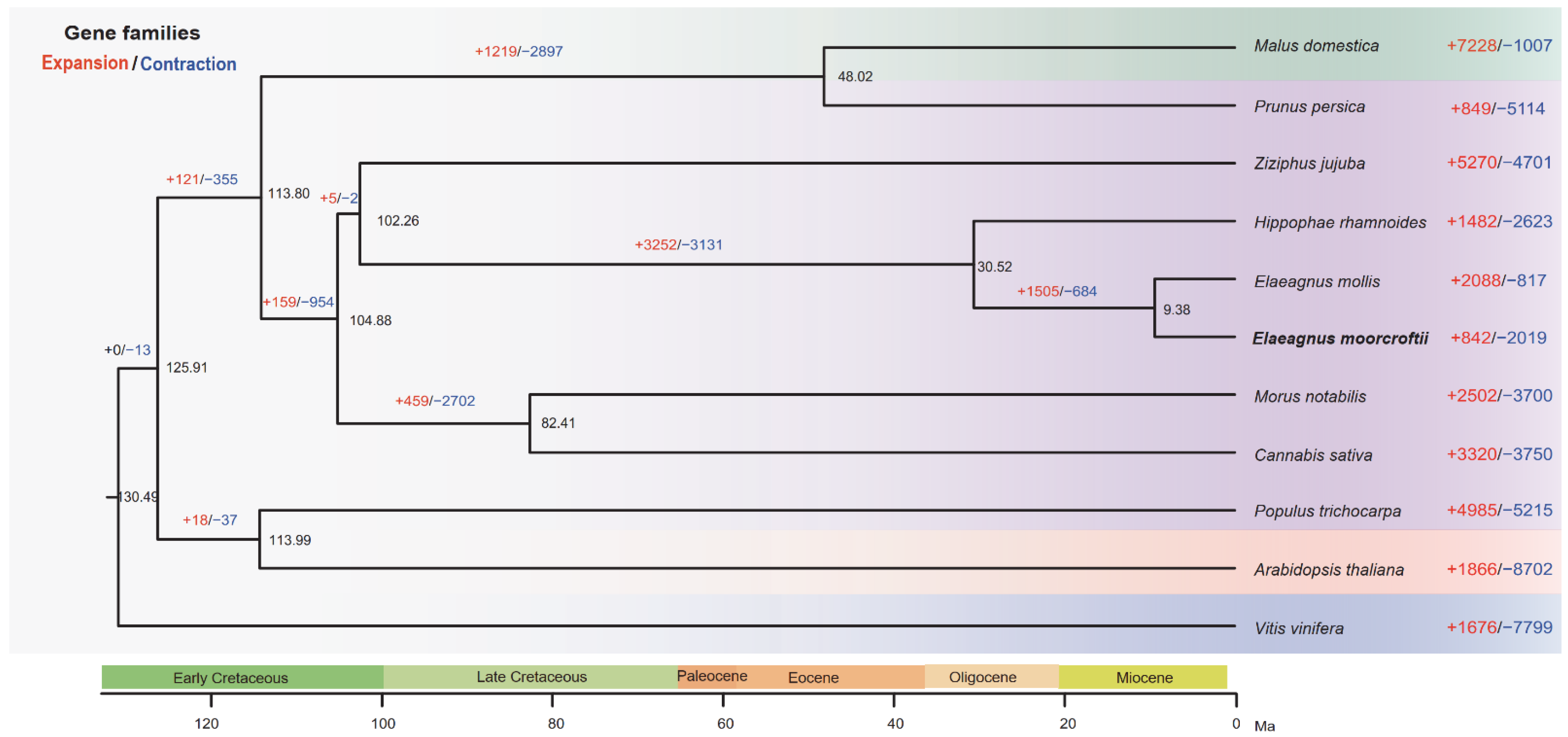
3.3. Evolutionary History of E. Moorcroftii
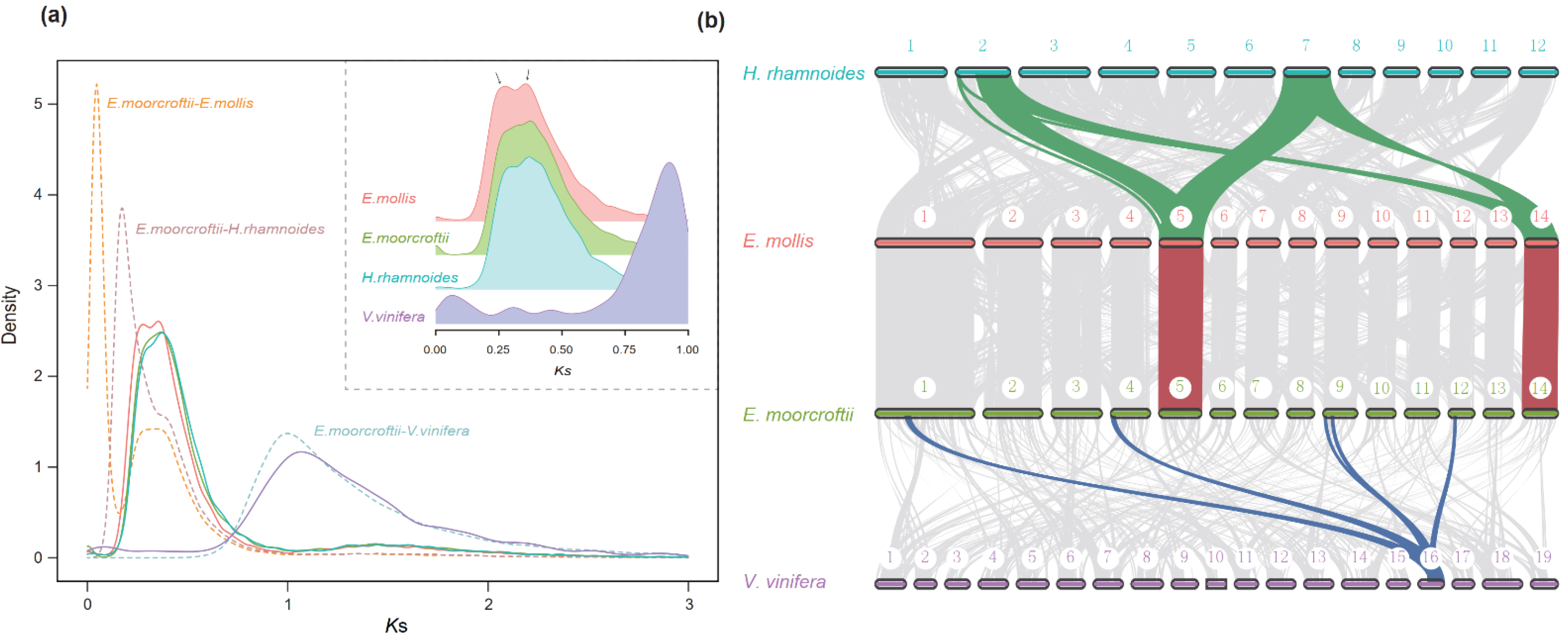
3.4. Whole-Genome Duplication Events in E. Moorcroftii
4. Discussion
4.1. A High-Quality Dryland Tree Species Genome
4.2. Differential Evolutionary Dynamics of Gene Families between E. Moorcroftii and E. Mollis
4.3. The Potential Evolutionary Significance of two Successive WGD Events in E. Moorcroftii
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reynolds, J.F.; Smith, D.M.S.; Lambin, E.F.; Turner, B.L., II; Mortimore, M.; Batterbury, S.P.J.; Downing, T.E.; Dowlatabadi, H.; Fernández, R.J.; Herrick, J.E.; et al. Global desertification: Building a science for dryland development. Science 2007, 316, 847–851. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yu, H.; Guan, X.; Wang, G.; Guo, R. Accelerated dryland expansion under climate change. Nat. Clim. Change 2016, 6, 166–172. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, G.; Zhang, Y.; Guan, X.; Wei, Y.; Guo, R. Global desertification vulnerability to climate change and human activities. Land Degrad. Dev. 2020, 31, 1380–1391. [Google Scholar] [CrossRef]
- Wu, Z.; Chen, H.; Pan, Y.; Feng, H.; Fang, D.; Yang, J.; Wang, Y.; Yang, J.; Sahu, S.K.; Liu, J.; et al. The genome of Hippophae rhamnoides provides insights into a conserved molecular mechanism in actinorhizal and rhizobial symbiosis. New Phytol. 2022, 235, 276–291. [Google Scholar] [CrossRef]
- Yu, L.; Diao, S.; Zhang, G.; Yu, J.; Zhang, T.; Luo, H.; Duan, A.; Wang, J.; He, C.; Zhang, J. Genome sequence and population genomics provide insights into chromosomal evolution and phytochemical innovation of Hippophae rhamnoides. Plant Biotechnol. J. 2022. [Google Scholar] [CrossRef]
- Wang, T.; Ren, L.; Li, C.; Zhang, D.; Zhang, X.; Zhou, G.; Gao, D.; Chen, R.; Chen, Y.; Wang, Z.; et al. The genome of a wild Medicago species provides insights into the tolerant mechanisms of legume forage to environmental stress. BMC Biol. 2021, 19, 96. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, L.; Tong, S.; Jiang, D.; Fu, Z. Chromosome-level genome assembly of a xerophytic plant, Haloxylon ammodendron. DNA Res. 2022, 29, dsac006. [Google Scholar] [CrossRef]
- Ma, W.; Wang, P.; Huang, J.; Zhang, D.; Yang, W.; Pan, B.; Shi, W. The taxonomic relationship between Elaeagnus moorcroftii and E. angustifolia (Elaeagnaceae) based on morphological similarities and simple-sequence repeat markers. Nord. J. Bot. 2021, 39, e03011. [Google Scholar] [CrossRef]
- Kucho, K.; Yamanaka, T.; Sasakawa, H.; Mansour, S.R.; Uchiumi, T. Different dynamics of genome content shuffling among host-specificity groups of the symbiotic actinobacterium Frankia. BMC Genom. 2014, 15, 609. [Google Scholar] [CrossRef]
- Liang, B.B.; Wang, W.J.; Fan, X.X.; Kurakov, A.V.; Liu, Y.F.; Song, F.Q.; Chang, W. Arbuscular mycorrhizal fungi can ameliorate salt stress in Elaeagnus angustifolia by improving leaf photosynthetic function and ultrastructure. Plant Biol. 2021, 23, 232–241. [Google Scholar] [CrossRef]
- Nazir, N.; Zahoor, M.; Nisar, M. A review on traditional uses and pharmacological importance of genus Elaeagnus species. Bot. Rev. 2020, 86, 247–280. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, P.; Wang, Y.L.; Zhang, Y. Nutritional composition of wild Elaeagnus angustifolia fruits. J. Gansu Agric. Univ. 2006, 41, 130–132. [Google Scholar]
- Ren, B.; Ru, D.; Chen, L.; Duan, N.; Li, Y.; Shi, J.; Cao, J.; Liu, B. Genome Sequence of Elaeagnus mollis, the first chromosome-level genome of the family Elaeagnaceae. Genome Biol. Evol. 2021, 13, evab266. [Google Scholar] [CrossRef] [PubMed]
- Allen, G.C.; Flores-Vergara, M.A.; Krasynanski, S.; Kumar, S.; Thompson, W.F. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat. Protoc. 2006, 1, 2320–2325. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef]
- Dudchenko, O.; Batra, S.S.; Omer, A.D.; Nyquist, S.K.; Hoeger, M.; Durand, N.C.; Shamim, M.S.; Machol, I.; Lander, E.S.; Aiden, A.P.; et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 2017, 356, 92–95. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with singlecopy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L. GMATA: An integrated software package for genome-scale SSR mining, marker development and viewing. Front Plant Sci. 2016, 7, 1350. [Google Scholar] [CrossRef]
- Gary, B. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 2, 573–580. [Google Scholar]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 2009, 25, 4.10.1–4.10.14. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Wessler, S.R. MITE-Hunter: A program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 2010, 38, e199. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. REPEATMODELER2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Keilwagen, J.; Wenk, M.; Erickson, J.L.; Schattat, M.H.; Grau, J.; Hartung, F. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 2016, 44, e89. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, C.; Zhao, X.; Fei, Z.; Wan, K.; Zhang, Z.; Pang, X.; Yin, X.; Bai, Y.; Sun, X. The Jujube genome provides insights into genome evolution and the domestication of sweetness/acidity taste in fruit trees. PLoS Genet. 2016, 12, e1006433. [Google Scholar] [CrossRef]
- Kaul, S.; Koo, H.L.; Jenkins, J.; Rizzo, M.; Rooney, T.; Tallon, L.J.; Feldblyum, T.; Nierman, W.; Benito, M.-I.; Lin, X.; et al. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815. [Google Scholar]
- Jaillon, O.; Aury, J.M.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.A.; Haas, B.J.; Hamilton, J.P.; Mount, S.M.; Buell, C.R. Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genom. 2006, 7, 327. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Bateman, A.; Marshall, M.; Khanna, A.; Eddy, S.R. RFAM: An RNA family database. Nucleic Acids Res. 2003, 31, 439–441. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 1–14. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software v.7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Shi, C.; Wang, S.; Cai, H.H.; Zhang, H.R.; Long, X.X.; Tihelka, E.; Song, W.; Feng, Q.; Jiang, R.; Cai, C.; et al. Fire-prone Rhamnaceae with South African affinities in Cretaceous Myanmar amber. Nat. Plants 2022, 8, 125–135. [Google Scholar] [CrossRef]
- De Bie, T.; Cristianini, N.; Demuth, J.P.; Hahn, M.W. CAFE: A computational tool for the study of gene family evolution. Bioinformatics 2006, 22, 1269–1271. [Google Scholar] [CrossRef]
- Qiao, X.; Li, Q.; Yin, H.; Qi, K.; Li, L.; Wang, R.; Zhang, S.; Paterson, A.H. Gene duplication and evolution in recurring polyploidization-diploidization cycles in plants. Genome Biol. 2019, 20, 38. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986, 3, 418. [Google Scholar]
- Jiao, Y.; Leebens-Mack, J.; Ayyampalayam, S.; Bowers, J.E.; McKain, M.R.; McNeal, J.; Rolf, M.; Ruzicka, D.R.; Wafula, E.; Wickett, N.J.; et al. A genome triplication associated with early diversification of the core eudicots. Genome Biol. 2012, 13, R3. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Zhou, G.; Yue, Z.; Hu, Q.; Chen, Y.; Liu, B.; Qiu, Q.; Wang, Z.; Zhang, J.; Wang, K.; et al. Genomic insights into salt adaptation in a desert poplar. Nat. Commun. 2014, 4, 2797. [Google Scholar] [CrossRef]
- Jiao, Y.; Wickett, N.J.; Ayyampalayam, S.; Chanderbali, A.S.; Landherr, L.; Ralph, P.E.; Tomsho, L.P.; Hu, Y.; Liang, H.; Soltis, P.S.; et al. Ancestral polyploidy in seed plants and angiosperms. Nature 2011, 473, 97–100. [Google Scholar] [CrossRef]
- Wang, J.; Sun, P.; Li, Y.; Liu, Y.; Yang, N.; Yu, J.; Ma, X.; Sun, S.; Xia, R.; Liu, X.; et al. An overlooked paleotetraploidization in Cucurbitaceae. Mol. Biol. Evol. 2018, 35, 16–26. [Google Scholar] [CrossRef]
- Soltis, P.S.; Soltis, D.E. Ancient WGD events as drivers of key innovations in angiosperms. Curr. Opin. Plant Biol. 2016, 30, 159–165. [Google Scholar] [CrossRef]
- Van de Peer, Y.; Mizrachi, E.; Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 2017, 18, 411–424. [Google Scholar] [CrossRef]
- Wu, S.; Han, B.; Jiao, Y. Genetic Contribution of Paleopolyploidy to Adaptive Evolution in Angiosperms. Mol. Plant 2020, 13, 59–71. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, S.; Li, L.; Yang, T.; Dong, S.; Wei, T.; Wu, S.; Liu, Y.; Gong, Y.; Feng, X.; et al. The Cycas genome and the early evolution of seed plants. Nat. Plants 2022, 8, 389–401. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Assembly | |
| Length of genome assembly (Mb) | 529.56 |
| Anchored to chromosome (Mb) | 500.73 |
| Contig N50 (Mb) | 28.21 |
| Longest contig (Mb) | 101.76 |
| BUSCO score of assembly (%) | 96.7% |
| Annotation | |
| GC content | 30.39% |
| Percentage of repeat sequences (%) | 60.95% |
| Number of protein-coding gene (%) | 29243 |
| Average gene length (bp) | 4318.92 |
| Average exon length (bp) | 220.21 |
| BUSCO score of annotation (%) | 98.5% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, X.; Wu, J.; Ma, X.; Li, K.; Zhang, H.; Wu, S.; Sun, K. The Chromosome-Level Genome of Elaeagnus moorcroftii Wall., an Economically and Ecologically Important Tree Species in Drylands. Diversity 2022, 14, 468. https://doi.org/10.3390/d14060468
Fu X, Wu J, Ma X, Li K, Zhang H, Wu S, Sun K. The Chromosome-Level Genome of Elaeagnus moorcroftii Wall., an Economically and Ecologically Important Tree Species in Drylands. Diversity. 2022; 14(6):468. https://doi.org/10.3390/d14060468
Chicago/Turabian StyleFu, Xinxing, Jingjing Wu, Xiaohui Ma, Kunpeng Li, Hui Zhang, Shengdan Wu, and Kun Sun. 2022. "The Chromosome-Level Genome of Elaeagnus moorcroftii Wall., an Economically and Ecologically Important Tree Species in Drylands" Diversity 14, no. 6: 468. https://doi.org/10.3390/d14060468
APA StyleFu, X., Wu, J., Ma, X., Li, K., Zhang, H., Wu, S., & Sun, K. (2022). The Chromosome-Level Genome of Elaeagnus moorcroftii Wall., an Economically and Ecologically Important Tree Species in Drylands. Diversity, 14(6), 468. https://doi.org/10.3390/d14060468