
Full-Length Transcriptome Sequencing Analysis and Characterization of Gene Isoforms Involved in Flavonoid Biosynthesis in the Seedless Kiwifruit Cultivar ‘Chengxiang’ (Actinidia arguta)

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and RNA Extraction
2.2. PacBio SMRT Library Construction and Sequencing
2.3. Functional Annotation and Classification
2.4. Prediction of Long Non-Coding RNAs (LncRNAs), Transcription Factors (TFs), and Simple Sequence Repeats (SSR)
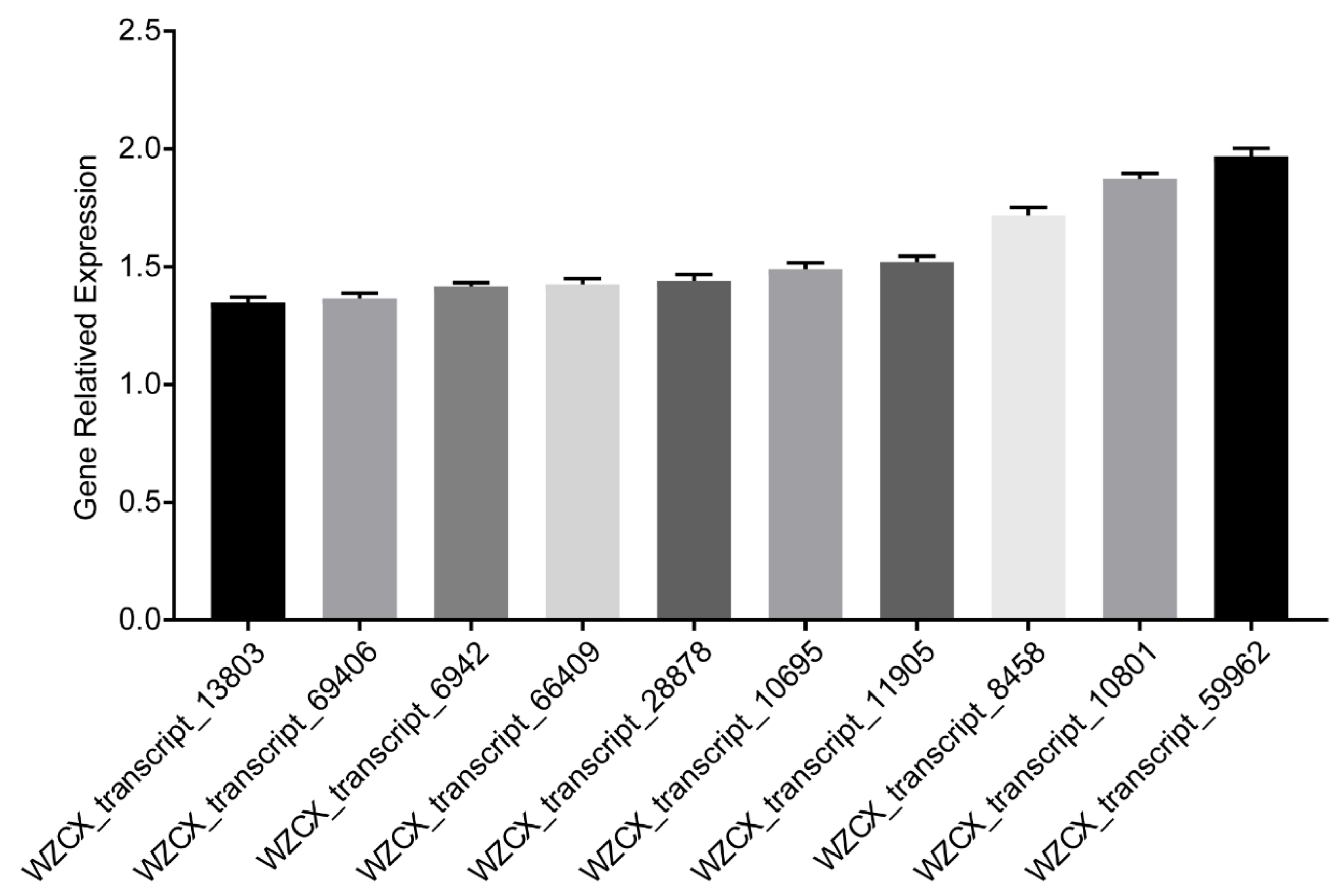
2.5. Gene Expression Analysis of Flavonoid Biosynthesis
2.6. Prediction of the CHS Family Members in the Seedless Kiwifruit Cultivar ‘Chengxiang’
2.7. Phylogenetic Analysis and Conserved Motif Analysis
3. Results
3.1. SMRT Sequencing of Kiwifruit Cultivar ‘Chengxiang’
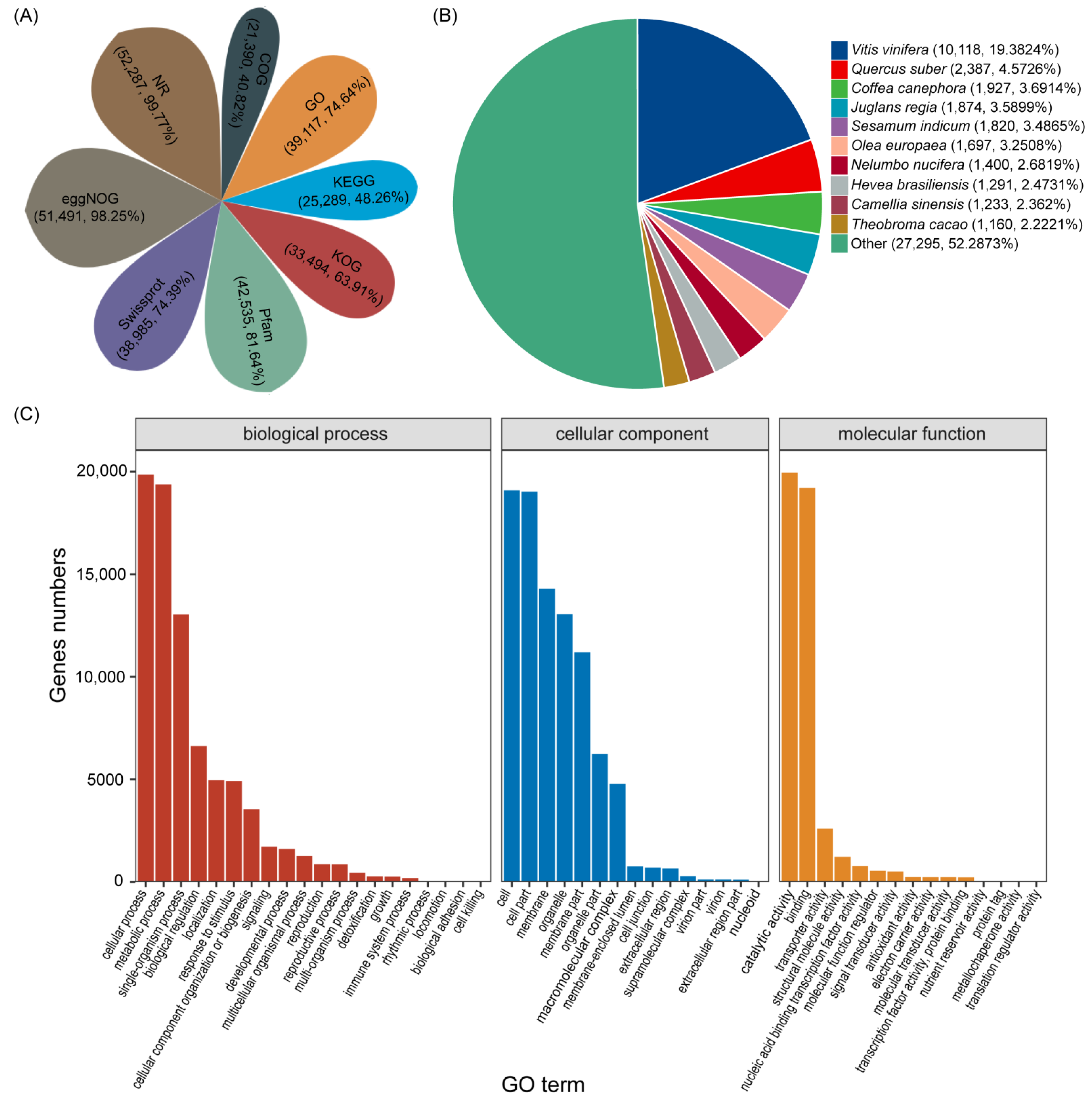
3.2. Functional Annotation of Transcripts
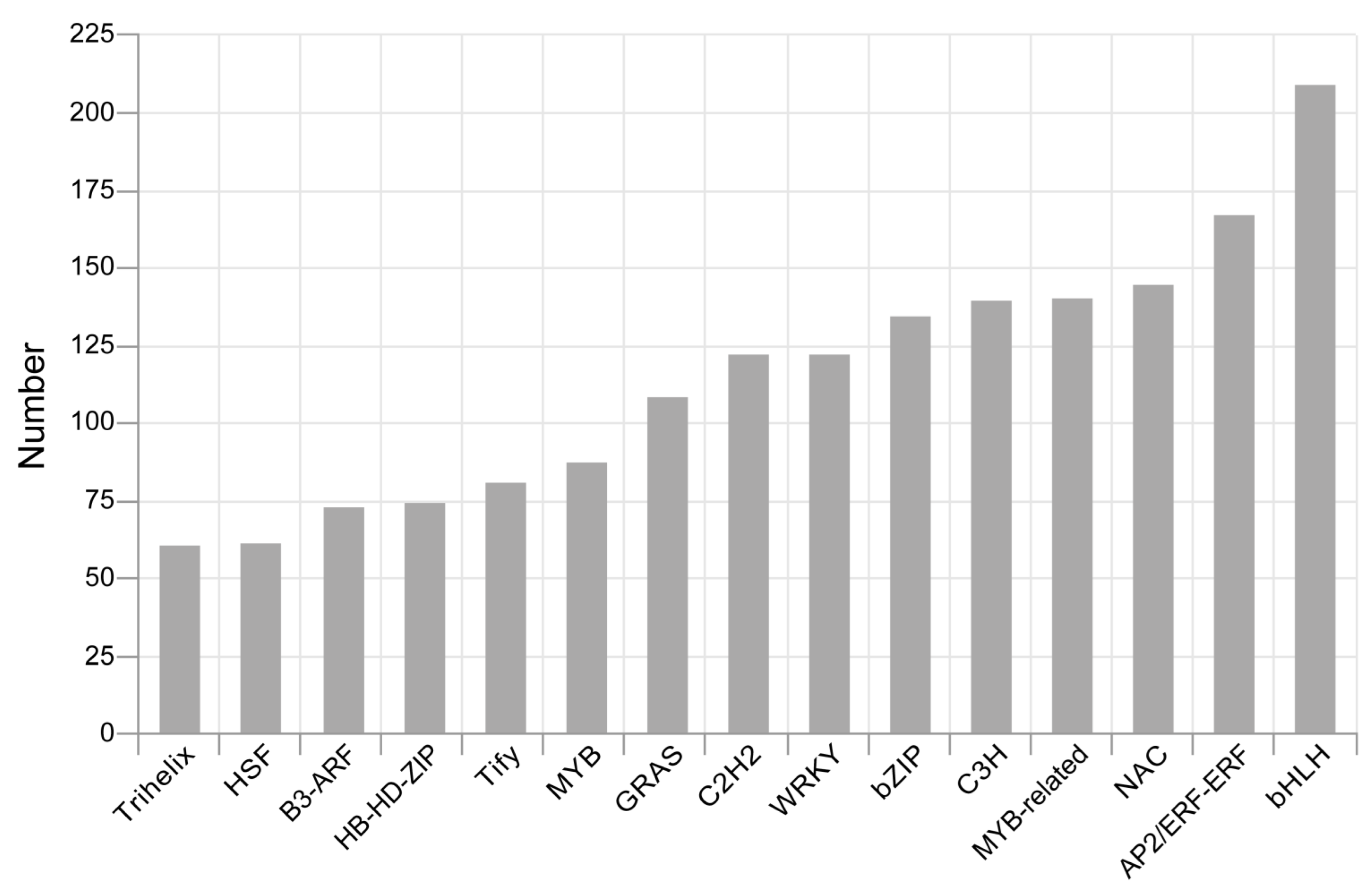
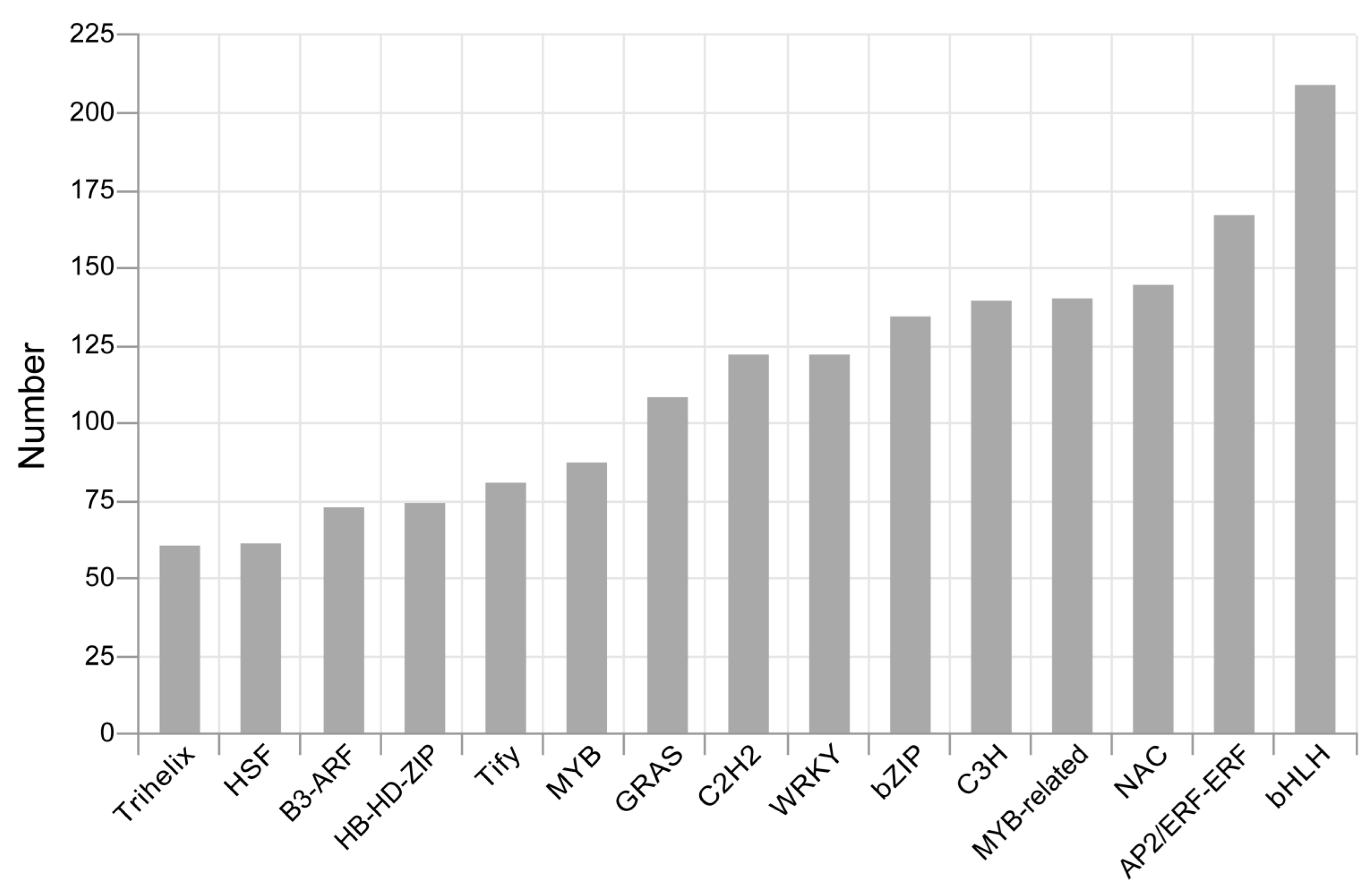
3.3. Identification of LncRNAs, TFs and SSRs
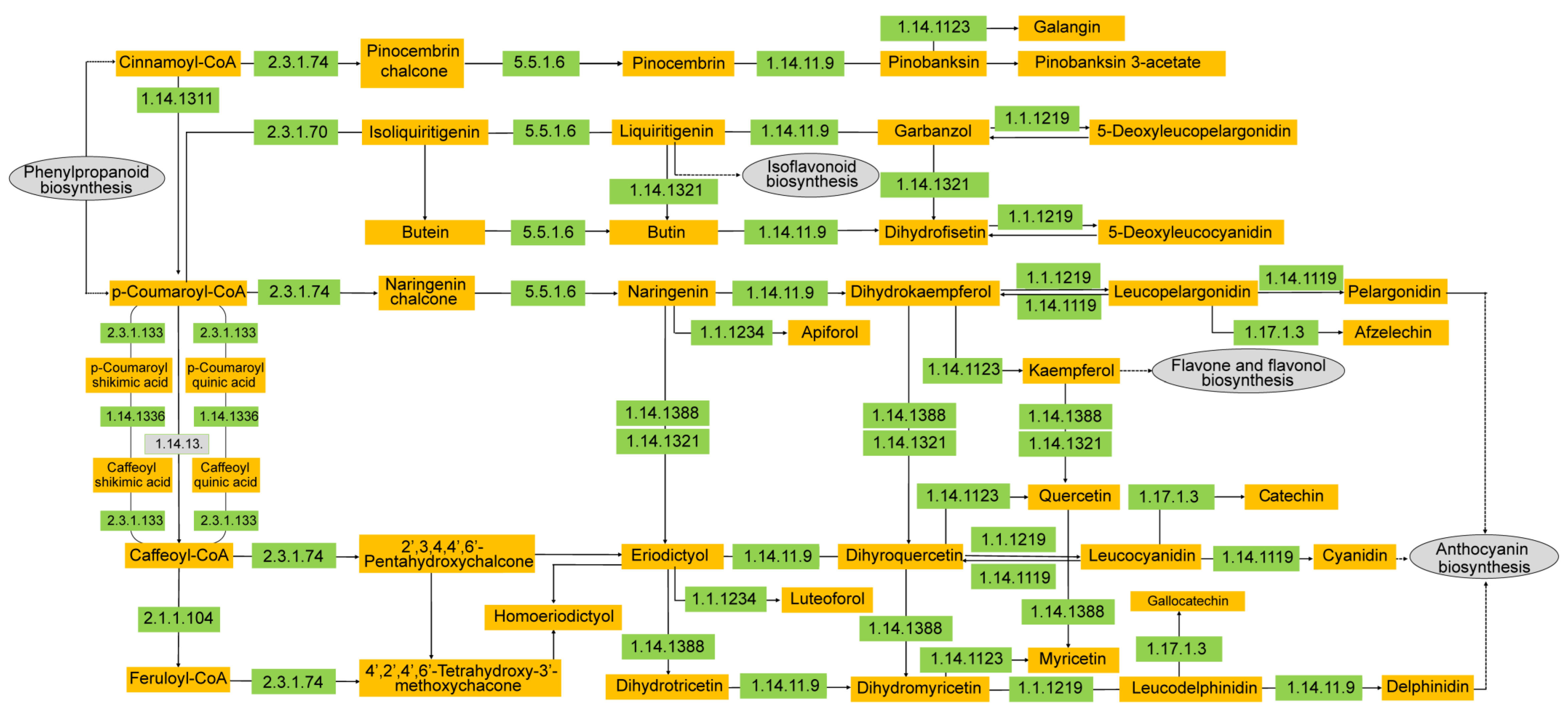
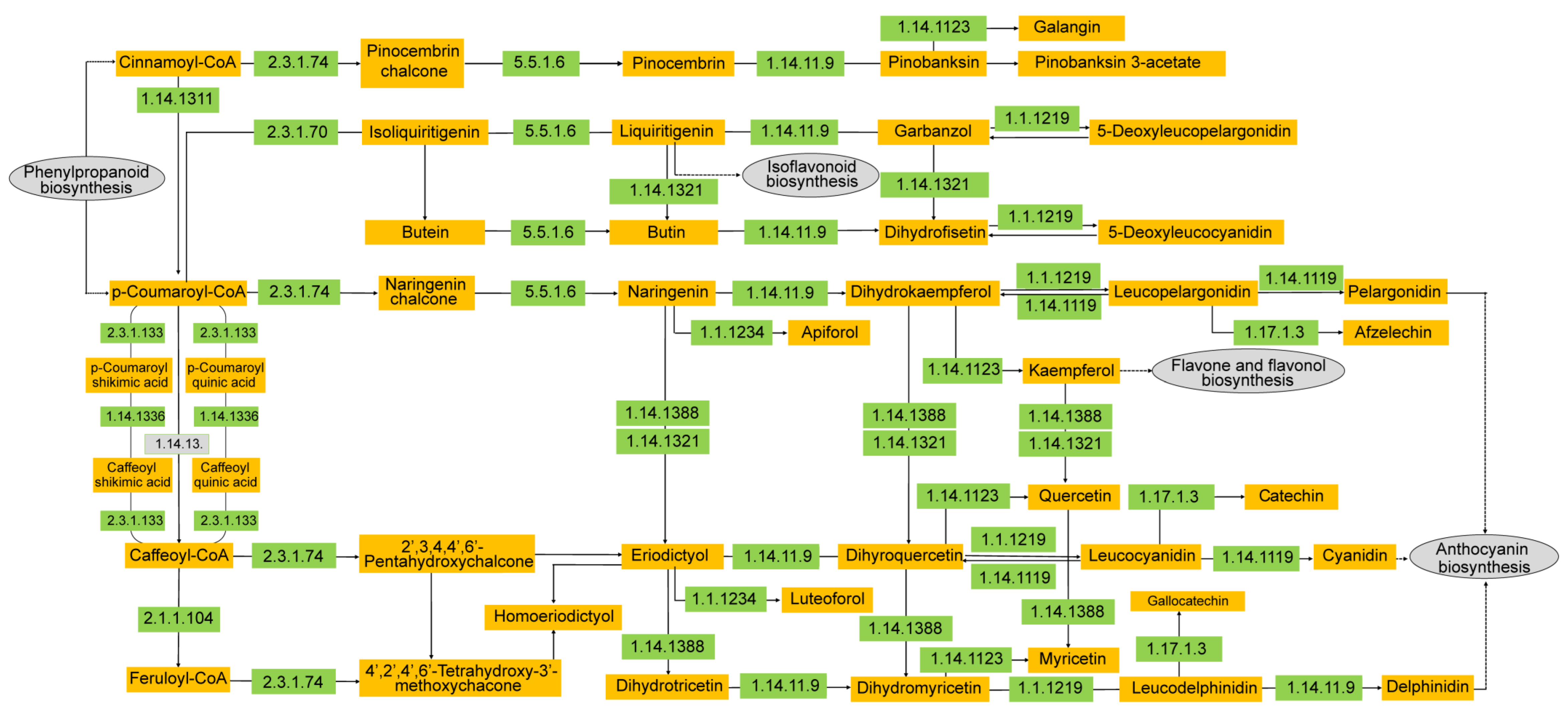
3.4. Candidate Genes Involved in Flavonoid Biosynthesis
3.5. Identification of the CHS Proteins in Seedless Kiwifruit Cultivar ‘Chengxiang’
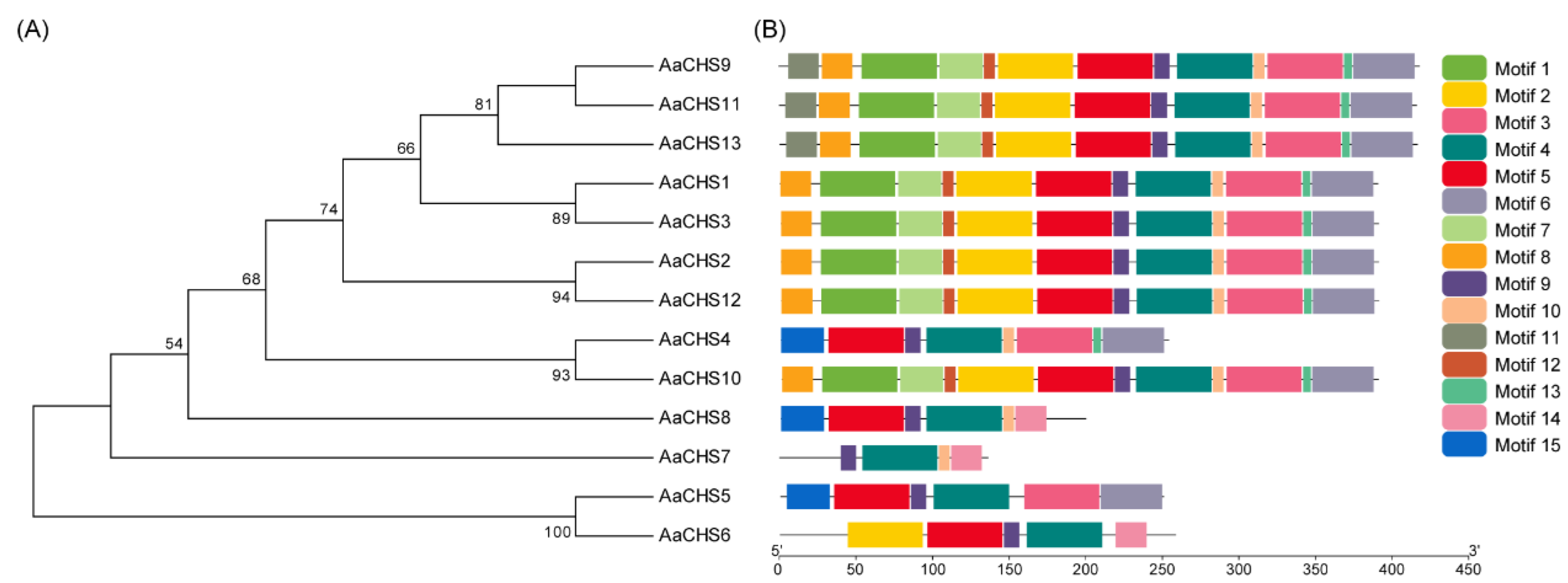
3.6. Phylogenetic Analyses and Motif Composition of CHS Gene Family in the Seedless Kiwifruit Cultivar ‘Chengxiang’ ,
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schijlen, E.G.W.M.; Ric de Vos, C.H.; Martens, S.; Jonker, H.H.; Rosin, F.M.; Molthoff, J.W.; Tikunov, Y.M.; Angenent, G.C.; van Tunen, A.J.; Bovy, A.G. RNA interference silencing of chalcone synthase, the first step in the flavonoid biosynthesis pathway, leads to parthenocarpic tomato fruits. Plant Physiol. 2007, 144, 1520–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingrosso, I.; Bonsegna, S.; Domenico, S.D.; Laddomada, B.; Blando, F.; Santino, A.; Giovinazzo, G. Over-expression of a grape stilbene synthase gene in tomato induces parthenocarpy and causes abnormal pollen development. Plant Physiol. Biochem. 2011, 49, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.J.; Shi, Q.C.; Albrecht, U.; Shatters Jr, R.G.; Stange, R.; McCollum, G.; Zhang, S.; Fan, C.M.; Stover, E. Comparative transcriptome analysis during early fruit development between three seedy citrus genotypes and their seedless mutants. Hortic. Res. 2017, 4, 17041. [Google Scholar] [CrossRef] [PubMed]
- Rotino, G.L.; Perri, E.; Zottini, M.; Sommer, H. Genetic engineering of parthenocarpic plants. Nat. Biotechnol. 1997, 15, 1398–1401. [Google Scholar] [CrossRef]
- Liu, W.; Chen, M.S.; Bai, L.J.; Zhuang, Z.H.; Fan, C.; Jiang, N.H.; Zhao, J.S.; Ma, S.P.; Xiang, X. Comprehensive transcriptomics and proteomics analyses of pollinated and parthenocarpic litchi (Litchi chinensis sonn.) fruits during early development. Sci. Rep. 2017, 7, 5401. [Google Scholar] [CrossRef]
- Varoquaux, F.; Blanvillain, R.; Delseny, M.; Gallois, P. Less is better: New approaches for seedless fruit production. Trends Biotechnol. 2000, 18, 233–242. [Google Scholar] [CrossRef]
- Gillaspy, G.; Ben-David, H.; Gruissem, W. Fruits: A developmental perspective. Plant Cell 1993, 5, 1439–1451. [Google Scholar] [CrossRef] [Green Version]
- Vivian-Smith, A.; Koltunow, A.M. Genetic analysis of growth-regulator-induced parthenocarpy in Arabidopsis. Plant Physiol. 1999, 121, 437–451. [Google Scholar] [CrossRef] [Green Version]
- Koes, R.; Verweij, W.; Quattrocchio, F. Flavonoids: A colourful model for the regulation and evolution of biochemical pathways. Trends Plant Sci. 2005, 10, 236–242. [Google Scholar] [CrossRef]
- Ylstra, B.; Touraev, A.; Benito Moreno, R.M.; Stoger, E.; van Tunen, A.J.; Vicente, O.; Mol, J.N.M.; Heberle-Bors, E. Flavonols stimulate development, germination and tube growth of tobacco pollen. Plant Physiol. 1992, 100, 902–907. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.L.; Ma, T.; Kang, M.H.; Ai, F.D.; Zhang, J.L.; Dong, G.Y.; Liu, J.Q. A high-quality Actinidia chinensis (kiwifruit) genome. Hortic. Res. 2019, 6, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Fang, J.; Qi, X.; Lin, M.; Zhong, Y.; Sun, L.; Cui, W. Combined analysis of the fruit metabolome and transcriptome reveals candidate genes involved in flavonoid biosynthesis in Actinidia arguta. Int. J. Mol. Sci. 2018, 19, 1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, D.; Pinto, D.; Santos, J.; Vinha, A.F.; Palmeira, J.; Ferreira, H.N.; Rodrigues, F.; Oliveira, M.B.P. Hardy kiwifruit leaves (Actinidia arguta): An extraordinary source of value-added compounds for food industry. Food Chem. 2018, 259, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fang, J.; Qi, X.; Lin, M.; Zhong, Y.; Sun, L. A key structural gene, AaLDOX, is involved in anthocyanin biosynthesis in all red-fleshed kiwifruit (Actinidia arguta) based on transcriptome analysis. Gene 2018, 648, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cui, W.; Wang, R.; Lin, M.; Fang, J. Microrna858-mediated regulation of anthocyanin biosynthesis in kiwifruit (Actinidia arguta) based on small RNA sequencing. PLoS ONE 2019, 14, e0217480. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.H.; Wang, Z.G.; Feng, X.L.; Pan, B.T.; Irfan, M.; Liu, C.J. Transcriptomic and metabolomics of flavonoid compounds in Actinidia arguta var. Arguta. J. King Saud Univ. Sci. 2021, 33, 101605. [Google Scholar] [CrossRef]
- Wojdyło, A.; Nowicka, P. Anticholinergic effects of Actinidia arguta fruits and their polyphenol content determined by liquid chromatography-photodiode array detector-quadrupole/time off light-mass spectrometry (LC-MS-PDA-Q/TOF). Food Chem. 2019, 271, 216–223. [Google Scholar] [CrossRef]
- Matich, A.J.; Young, H.; Allen, J.M.; Wang, M.Y.; Fielder, S.; McNeilage, M.A.; MacRae, E.A. Actinidia arguta: Volatile compounds in fruit and flowers. Phytochemistry 2003, 63, 285–301. [Google Scholar] [CrossRef]
- Jang, D.S.; Lee, G.Y.; Lee, Y.M.; Kim, Y.S.; Sun, H.; Kim, D.H.; Kim, J.S. Flavan-3-ols having a g-Lactam from the roots of Actinidia arguta inhibit the formation of advanced glycation end products in Vitro. Chem. Pharm. Bull. 2009, 57, 397–400. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Yang, M.; Yu, Z.; Zhang, T. Characterization of the whole transcriptome of whelk Rapana venosa by single-molecule mRNA sequencing. Mar. Genom. 2019, 44, 74–77. [Google Scholar] [CrossRef]
- Li, W.; Jaroszewski, L.; Godzik, A. Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 2002, 18, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, D412–D419. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Harris, M.A. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Salazar, G.A. The Pfam protein family’s database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.G.; Park, H.J.; Dasari, S.; Wang, S.Q.; Kocher, J.P.; Li, W. CPAT: Coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Jiao, W.; Song, H.; Qu, H.; Li, D.; Mei, H.; Tong, Q. miRNA-558 promotes gastric cancer progression through attenuating Smad4-mediated repression of heparanase expression. Cell Death Dis. 2016, 7, e2382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2DDCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Rédei, G.P. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, 202–208. [Google Scholar] [CrossRef]
- Nvh, A.; Rjh, B. Iso-Seq Long Read Transcriptome Sequencing. In Comprehensive Foodomics; Cifuentes, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 486–500. [Google Scholar]
- Wang, Z.B.; Yu, Q.B.; Shen, W.X.; El Mohtar, C.A.; Zhao, X.C.; Gmitter, F.G., Jr. Functional study of CHS gene family members in citrus revealed a novel chs gene affecting the production of flavonoids. BMC Plant Biol. 2018, 18, 189. [Google Scholar] [CrossRef] [PubMed]
- Li, W.B.; Liu, Y.F.; Zeng, S.H.; Xiao, G.; Wang, G.; Wang, Y.; Peng, M.; Huang, H.W. Gene expression profiling of development and anthocyanin accumulation in kiwifruit (Actinidia chinensis) based on transcriptome sequencing. PLoS ONE 2015, 10, e0136439. [Google Scholar]
- Zuo, C.; Matthew, B.; Avinash, S.; Kuo, R.C.; Kunde, R.G.; Torres-Jerez, I.; Li, G.F.; Wang, M.; Dilworth, D.; Barry, K.; et al. Revealing the transcriptomic complexity of switchgrass by pacbio long-read sequencing. Biotechnol. Biofuels 2018, 11, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Qiu, G.; Yang, M. SMRT sequencing of full-length transcriptome of seagrasses. Sci. Rep. 2019, 9, 14537. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef] [Green Version]
- Karlik, E.; Marakli, S.; Gozukirmizi, N. Two lncRNAs expression profiles in salt stressed barley (Hordeum vulgare L.) roots. Cytologia 2018, 83, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Lin, J.; Kan, J.; Wang, H.; Li, X.; Yang, Q.; Li, H.; Chang, Y. Genome-wide identification and functional prediction of novel drought-responsive LncRNAs in Pyrus betulifolia. Genes 2018, 9, 311. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Wu, Z.; Gu, H.; Cheng, D.; Guo, X.; Li, L.; Shi, C.; Xu, G.; Gu, S.; Abid, M.; et al. AvNAC030, a NAC Domain Transcription Factor, Enhances Salt Stress Tolerance in Kiwifruit. Int. J. Mol. Sci. 2021, 22, 11897. [Google Scholar] [CrossRef]
- Wang, R.C.; Shu, P.; Zhang, C.; Zhang, J.L.; Chen, Y.; Zhang, Y.X.; Du, K.; Xie, Y.; Li, M.Z.; Ma, T.; et al. Integrative analyses of metabolome and genome-widetranscriptome reveal the regulatory network governing flavor formation in kiwifruit (Actinidia chinensis). New Phytol. 2022, 233, 373–389. [Google Scholar] [CrossRef]
- Ampomah-Dwamena, C.; Thrimawithana, A.H.; Dejnoprat, S.; Lewis, D.; Espley, R.V.; Allan, A.C. A kiwifruit (Actinidia deliciosa) R2R3-MYB transcription factor modulates chlorophyll and carotenoid accumulation. New Phytol. 2019, 221, 309–325. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.Y.; Chen, X.R.; Wang, J.P.; Cui, W.Q.; Xing, X.X.; Chen, X.Y.; Ding, W.Y.; God’spower, B.O.; Eliphaz, N.; Sun, M.Q.; et al. Transcriptomic analysis reveals flavonoid biosynthesis of Syringa oblata Lindl. in response to different light intensity. BMC Plant Biol. 2019, 19, 487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agati, G.; Azzarello, E.; Pollastri, S.; Tattini, M. Flavonoids as antioxidants in plants: Location and functional significance. Plant Sci. 2012, 196, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuka, T.; Saito, M.; Yamada, E.; Fujita, K.; Kakizaki, Y.; Nishihara, M. Isolation and characterization of GtMYBP3 and GtMYBP4, orthologues of R2R3-MYB transcription factors that regulate early flavonoid biosynthesis, in gentian flowers. J. Exp. Bot. 2012, 63, 6505–6517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wani, T.A.; Pandith, S.A.; Gupta, A.P.; Chandra, S.; Sharma, N.; Lattoo, S. Molecular and functional characterization of two isoforms of chalcone synthase and their expression analysis in relation to flavonoid constituents in Grewia asiatica L. PLoS ONE 2017, 12, e0179155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos, É.; Hamdan, F.F. Cloning of the chaperonin t-complex polypeptide 1 gene from Schistosoma mansoni and studies of its expression levels under heat shock and oxidative stress. Parasitol. Res. 2000, 86, 253–258. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data | Platform | Number of Sequence |
|---|---|---|
| Circular Consensus (CCS) reads | SMRT | 161,390 |
| Number of full-length non-chimeric reads | SMRT | 145,210 |
| High quality consensus sequence | IsoSeq | 80,615 |
| Nonredundant transcripts | CD-HIT | 57,721 |
| Gene | Enzyme | Enzyme Code | KEGG ID | Transcripts |
|---|---|---|---|---|
| CHS | chalcone synthase | EC:2.3.1.74 | K00660 | 15 |
| CHI | chalcone isomerase | EC:5.5.1.6 | K01859 | 2 |
| FLS | flavonol synthase | EC:1.14.11.23 | K05278 | 12 |
| F3′5′H | flavonoid 3′,5′-hydroxylase | EC:1.14.13.88 | K13083 | 2 |
| DFR | bifunctional dihydroflavonol 4-reductase/flavanone 4-reductase | EC:1.1.1.219 1.1.1.234 | K13082 | 5 |
| ANS | anthocyanidin synthase | EC:1.14.11.19 | K05277 | 16 |
| F3H | naringenin 3-dioxygenase | EC:1.14.11.9 | K00475 | 8 |
| C4H | trans-cinnamate 4-monooxygenase | EC:1.14.13.11 | K00487 | 4 |
| F3′H | flavonoid 3′-monooxygenase | EC:1.14.13.21 | K05280 | 3 |
| F3′H1 | coumaroylquinate(coumaroylshikimate) 3′-monooxygenase | EC:1.14.13.36 | K09754 | 1 |
| LAR | leucoanthocyanidin reductase | EC:1.17.1.3 | K13081 | 7 |
| ANR | anthocyanidin reductase | EC:1.3.1.112 | K21102 | 3 |
| CoAOMT | caffeoyl-CoA O-methyltransferase | EC:2.1.1.104 | K00588 | 7 |
| HCT | shikimate O-hydroxycinnamoyltransferase | EC:2.3.1.133 | K13065 | 13 |
| Gene Name | Accession Number | Amino Acid | Molecular Weight (Da) | Isoelectric Points | Instability Index | Aliphatic Index |
|---|---|---|---|---|---|---|
| AaCHS1 | WZCX_transcript_10099 | 390 | 42,660.45 | 6.23 | 35.59 | 91 |
| AaCHS2 | WZCX_transcript_10244 | 390 | 42,604.35 | 6.18 | 35.86 | 92.49 |
| AaCHS3 | WZCX_transcript_19378 | 390 | 42,660.45 | 6.23 | 35.59 | 91 |
| AaCHS4 | WZCX_transcript_36023 | 253 | 27,030.28 | 5.81 | 29.58 | 103.69 |
| AaCHS5 | WZCX_transcript_43527 | 250 | 27,236.33 | 5.42 | 35.77 | 93.61 |
| AaCHS6 | WZCX_transcript_48357 | 258 | 27,916.1 | 5.05 | 49.8 | 96.07 |
| AaCHS7 | WZCX_transcript_48795 | 136 | 14,672.81 | 4.88 | 26.48 | 101.85 |
| AaCHS8 | WZCX_transcript_50909 | 199 | 21,321.53 | 6.21 | 33.18 | 97.07 |
| AaCHS9 | WZCX_transcript_65365 | 418 | 45,562.47 | 6.72 | 36.53 | 88.37 |
| AaCHS10 | WZCX_transcript_66409 | 389 | 42,499.24 | 6.73 | 36.81 | 91.21 |
| AaCHS11 | WZCX_transcript_69406 | 416 | 45,498.49 | 6.96 | 35.72 | 88.1 |
| AaCHS12 | WZCX_transcript_9529 | 390 | 42,662.39 | 6.04 | 36.08 | 92.49 |
| AaCHS13 | WZCX_transcript_9975 | 416 | 45,480.43 | 6.67 | 36.18 | 88.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, Y.; Wu, Y.-P.; Wang, F.-W.; Zhang, L.; Yu, G.; Wang, Y.-L.; Zhang, Y. Full-Length Transcriptome Sequencing Analysis and Characterization of Gene Isoforms Involved in Flavonoid Biosynthesis in the Seedless Kiwifruit Cultivar ‘Chengxiang’ (Actinidia arguta). Diversity 2022, 14, 424. https://doi.org/10.3390/d14060424
Jia Y, Wu Y-P, Wang F-W, Zhang L, Yu G, Wang Y-L, Zhang Y. Full-Length Transcriptome Sequencing Analysis and Characterization of Gene Isoforms Involved in Flavonoid Biosynthesis in the Seedless Kiwifruit Cultivar ‘Chengxiang’ (Actinidia arguta). Diversity. 2022; 14(6):424. https://doi.org/10.3390/d14060424
Chicago/Turabian StyleJia, Yun, Yong-Peng Wu, Feng-Wei Wang, Lei Zhang, Gang Yu, Ya-Ling Wang, and Ying Zhang. 2022. "Full-Length Transcriptome Sequencing Analysis and Characterization of Gene Isoforms Involved in Flavonoid Biosynthesis in the Seedless Kiwifruit Cultivar ‘Chengxiang’ (Actinidia arguta)" Diversity 14, no. 6: 424. https://doi.org/10.3390/d14060424
APA StyleJia, Y., Wu, Y.-P., Wang, F.-W., Zhang, L., Yu, G., Wang, Y.-L., & Zhang, Y. (2022). Full-Length Transcriptome Sequencing Analysis and Characterization of Gene Isoforms Involved in Flavonoid Biosynthesis in the Seedless Kiwifruit Cultivar ‘Chengxiang’ (Actinidia arguta). Diversity, 14(6), 424. https://doi.org/10.3390/d14060424