
Tracking the Origins of Pseudomonas aeruginosa Phylogroups by Diversity and Evolutionary Analysis of Important Pathogenic Marker Genes †

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Analysis of Genome Sequences
2.2. Determination of Average Nucleotide Identity (ANI) Index among P. aeruginosa Genomes
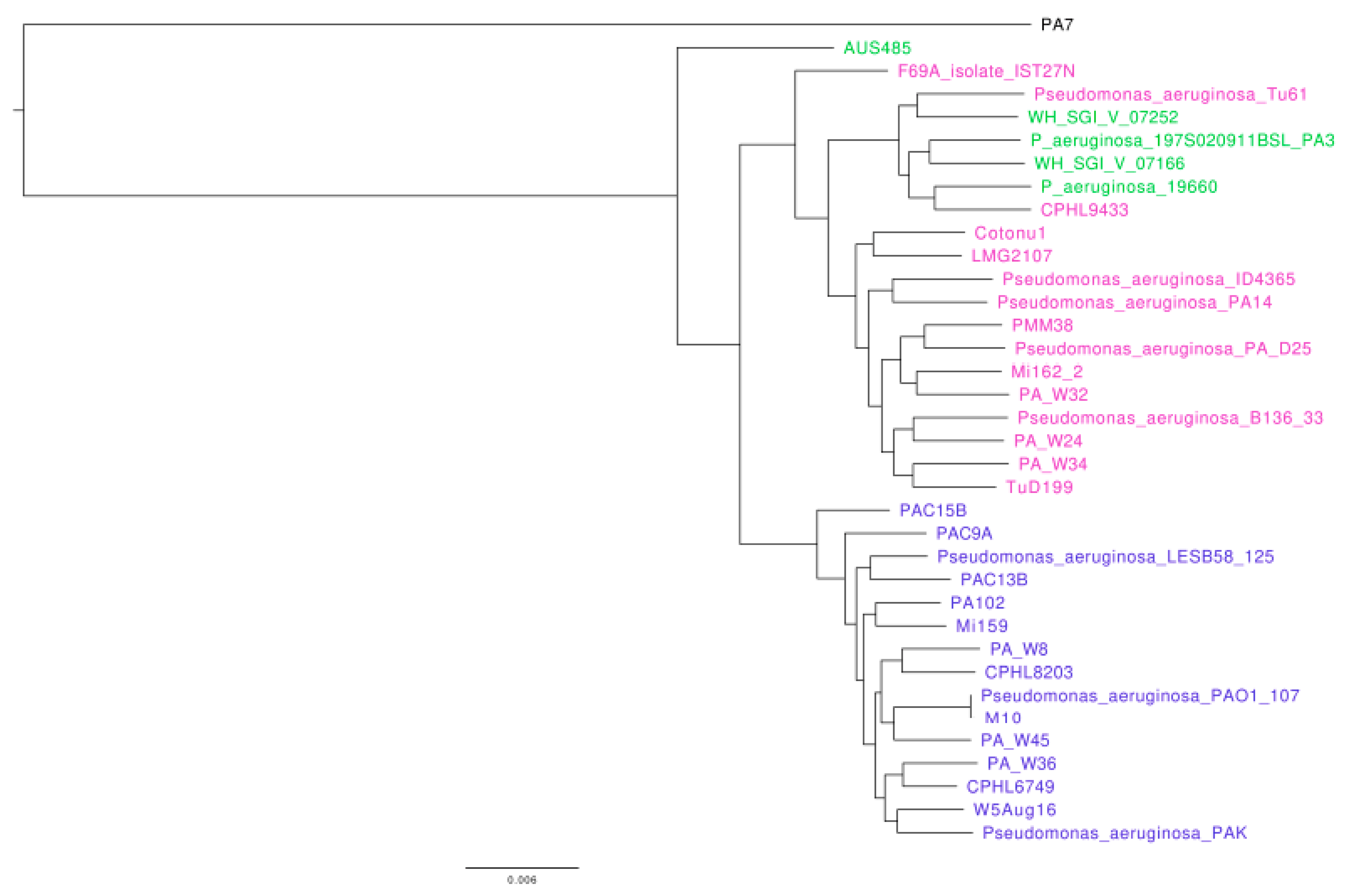
2.3. Construction of Phylogenetic Trees
3. Results and Discussion
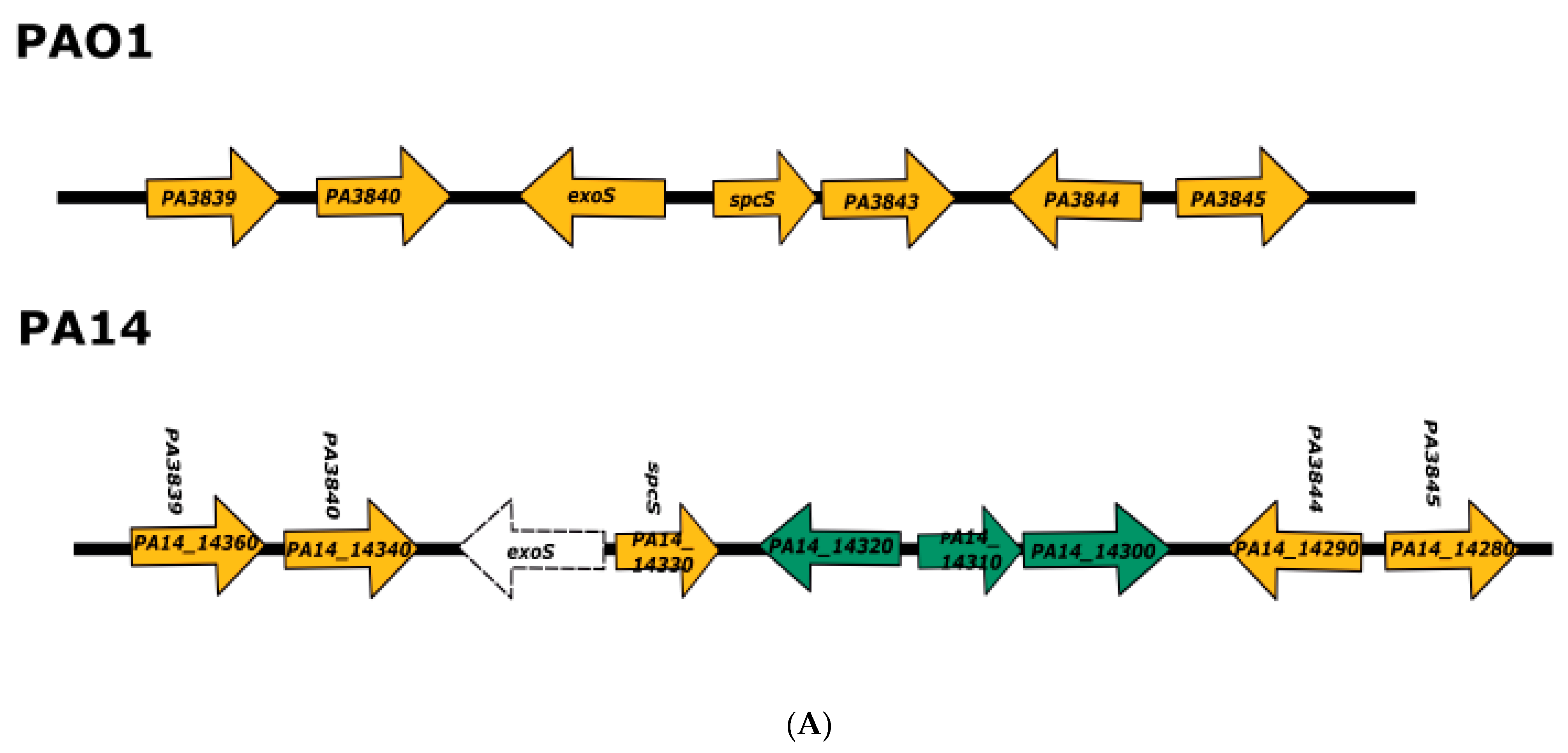
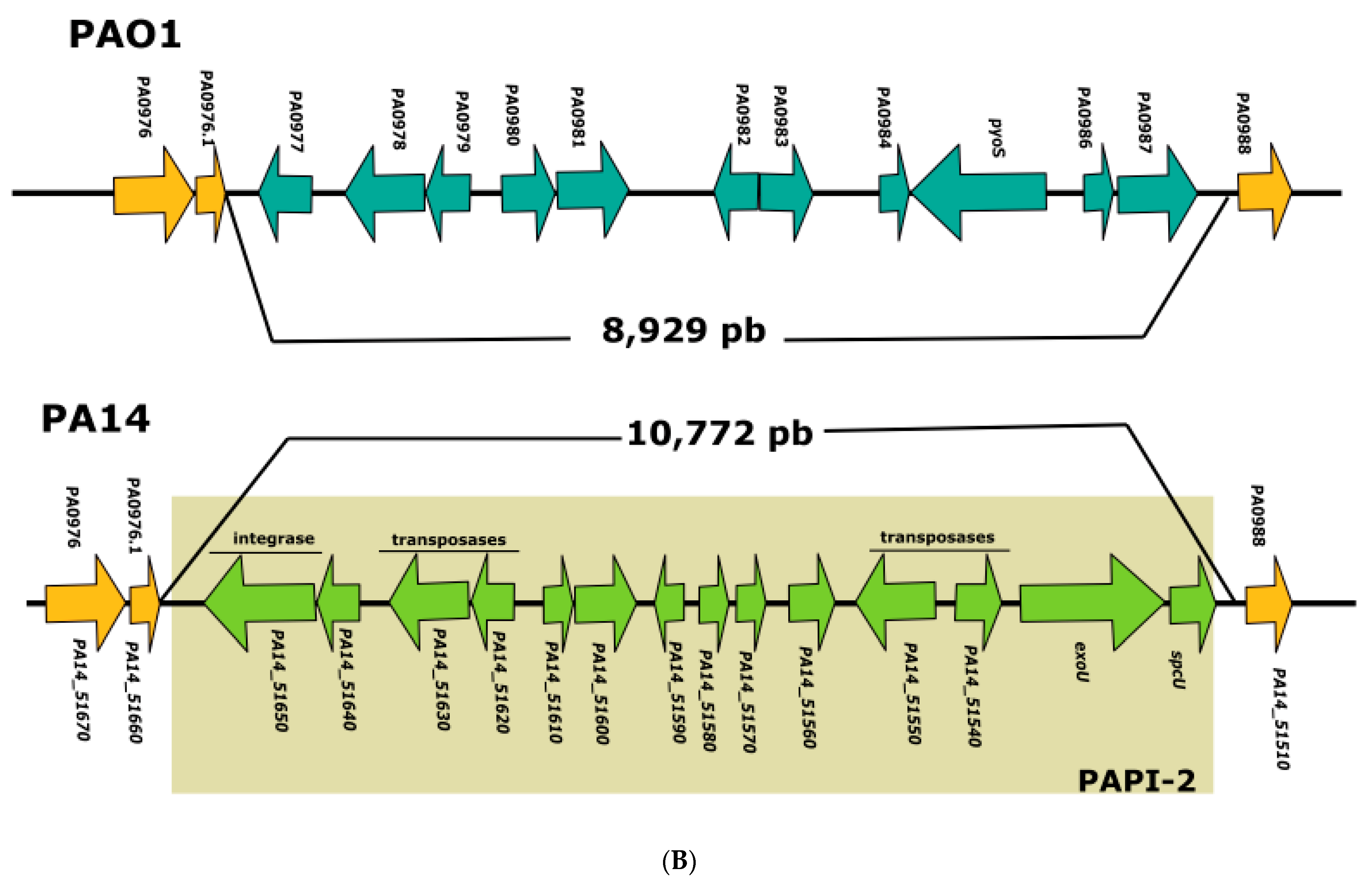
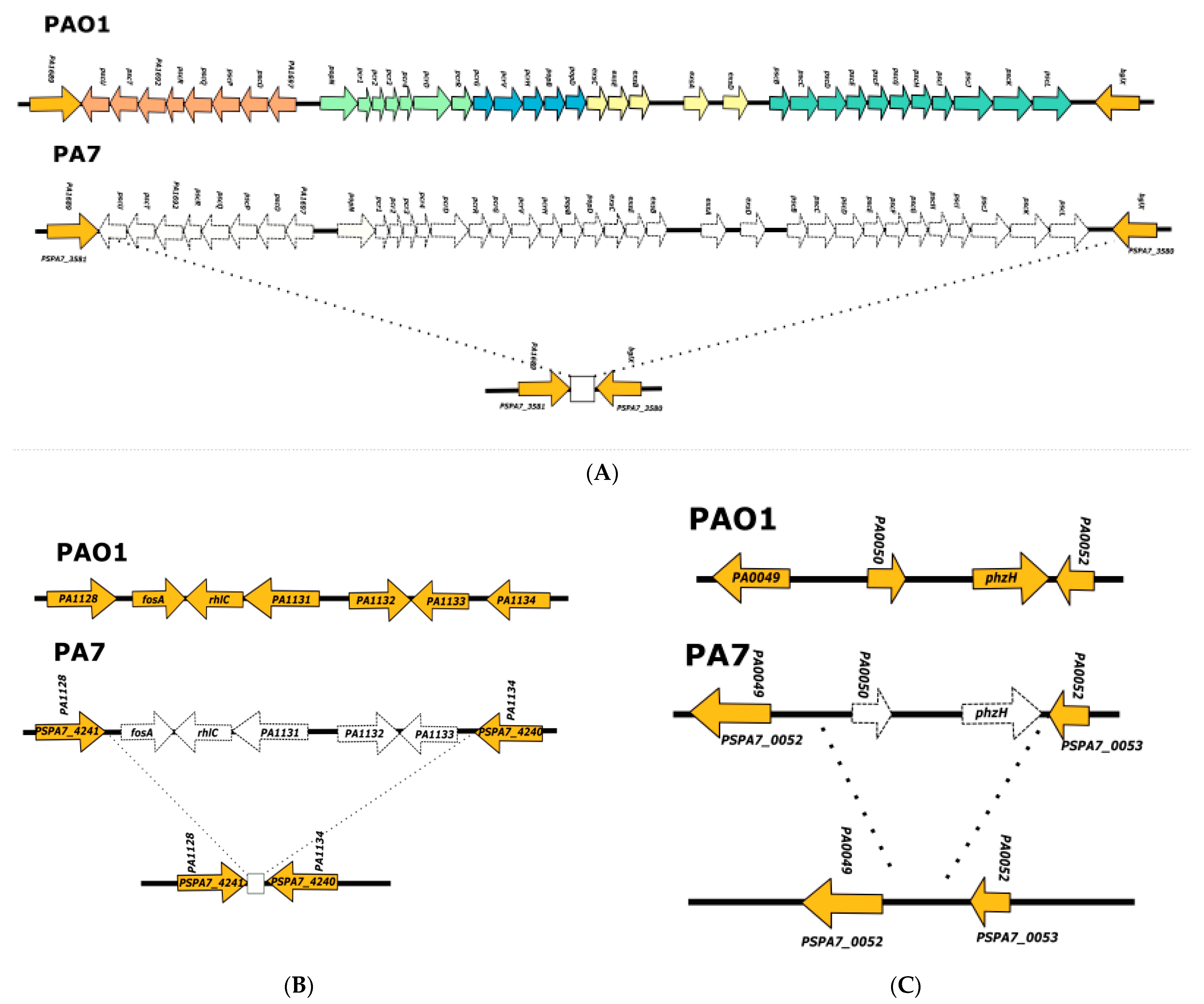
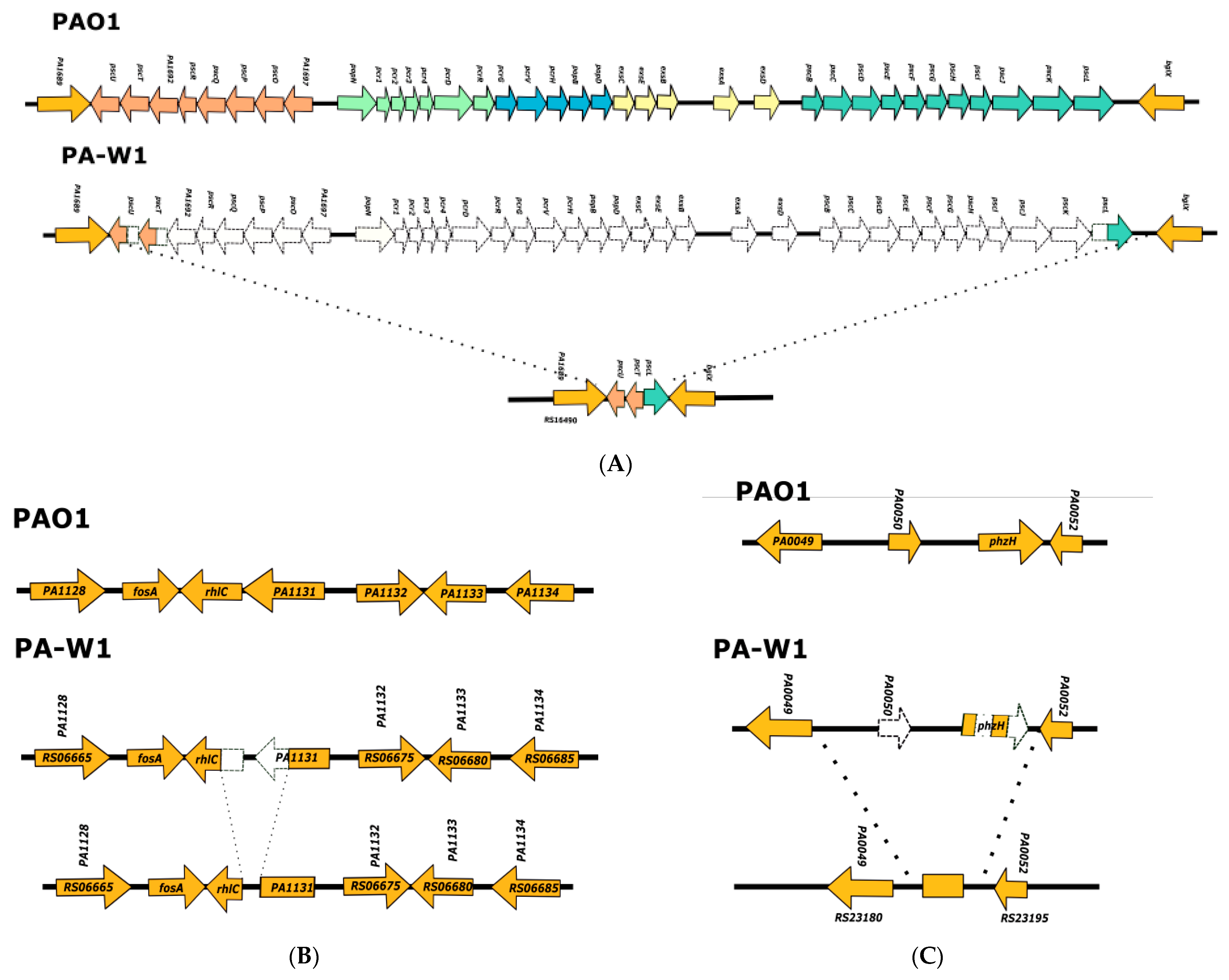
3.1. Analysis of the Genomes of Strains Belonging to Clades 1 and 2
3.2. Analysis of the Genomes of Strains Belonging to Clade 3
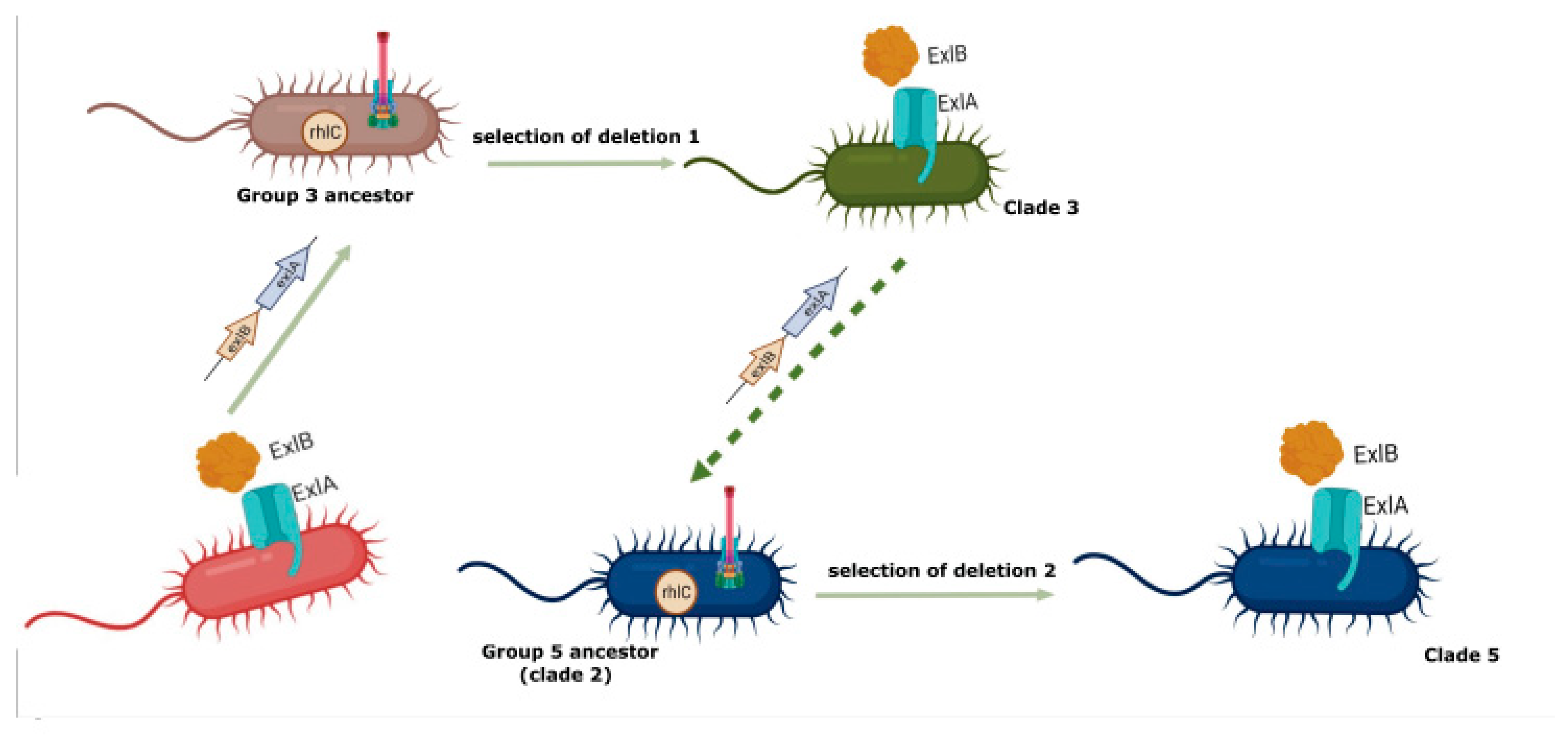
3.3. Analysis of the Genomes of Strains Belonging to Clade 5
3.4. Searching for Clade 4
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moradali, M.F.; Ghods, S.; Rehm, B.H.A. Pseudomonas aeruginosa lifestyle: A paradigm for adaptation, survival and persistence. Front. Cell Infect. Microbiol. 2017, 7, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crone, S.; Vives-Flórez, M.; Kvich, L.; Saunders, A.M.; Malone, M.; Nicolaisen, M.H.; Martínez-García, E.; Rojas-Acosta, C.; Gomez-Puerto, M.C.; Calum, H.; et al. The environmental occurrence of Pseudomonas aeruginosa. APMIS 2020, 128, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Diggle, S.P.; Whiteley, M. Microbe Profile: Pseudomonas aeruginosa: Opportunistic pathogen and lab rat. Microbiol.-SGM 2020, 166, 30–33. [Google Scholar] [CrossRef]
- Jurado-Martín, I.; Sainz-Mejías, M.; McClean, S. Pseudomonas aeruginosa: An audacious pathogen with an adaptable arsenal of virulence factors. Int. J. Mol. Sci. 2021, 22, 3128. [Google Scholar] [CrossRef]
- Gellatly, S.L.; Hancock, R.E.W. Pseudomonas aeruginosa: New insights into pathogenesis and host defenses. Pathog. Dis. 2013, 67, 159–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, Z.; Raudonis, R.; Glick, B.R.; Lin, T.J.; Cheng, Z. Antibiotic resistance in Pseudomonas aeruginosa: Mechanisms and alternative therapeutic strategies. Biotechnol. Adv. 2019, 37, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.R.; Shrivastava, P.S.; Ramasamy, J. Responding to the challenge of antibiotic resistance: World Health Organization. J. Res. Med. Sci. 2018, 23, 21. [Google Scholar] [CrossRef]
- Wolfgang, M.C.; Kulasekara, B.R.; Liang, X.; Boyd, D.; Wu, K.; Yang, Q.; Miyada, C.G.; Lory, S. Conservation of genome content and virulence determinants among clinical and environmental isolates of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2003, 100, 8484–8489. [Google Scholar] [CrossRef] [Green Version]
- Grosso-Becerra, M.V.; Santos-Medellín, C.; González-Valdez, A.; Méndez, J.L.; Delgado, G.; Morales-Espinosa, R.; Servín-González, L.; Alcaraz, L.-D.; Soberón-Chávez, G. Pseudomonas aeruginosa clinical and environmental isolates constitute a single population with high phenotypic diversity. BMC Genom. 2014, 15, 318. [Google Scholar] [CrossRef] [Green Version]
- Williams, P.; Cámara, M. Quorum sensing and environmental adaptation in Pseudomonas aeruginosa: A tale of regulatory networks and multifunctional signal molecules. Curr. Opin. Microbiol. 2009, 12, 182–191. [Google Scholar] [CrossRef]
- Berube, B.J.; Murphy, K.R.; Torhan, M.C.; Bowlin, N.O.; Williams, J.D.; Bowlin, T.L.; Moir, D.T.; Hauser, A.R. Impact of type III secretion effectors and of phenoxyacetamide inhibitors of type III secretion on abscess formation in a mouse model of Pseudomonas aeruginosa infection. Antimicrob. Agents Chemother. 2017, 61, e01202–e01217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilker, R.; Munder, A.; Klockgether, J.; Losada, P.M.; Chouvarine, P.; Cramer, N.; Davenport, C.F.; Dethlefsen, S.; Fischer, S.; Peng, H.; et al. Interclonal gradient of virulence in the Pseudomonas aeruginosa pangenome from disease and environment. Environ. Microbiol. 2015, 17, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.H.; Tetu, S.G.; Larouche, A.; Elbourne, L.; Tremblay, S.; Ren, Q.; Dodson, R.; Harkins, D.; Shay, R.; Watkins, K.; et al. Complete genome sequence of the multiresistant taxonomic outlier Pseudomonas aeruginosa PA7. PLoS ONE 2010, 5, e8842. [Google Scholar] [CrossRef] [PubMed]
- Freschi, L.; Jeukens, J.; Kukavica-Ibrulj, I.; Boyle, B.; Dupont, M.J.; Laroche, J.; Larose, S.; Maaroufi, H.; Fothergill, J.L.; Moore, M.; et al. Clinical utilization of genomics data produced by the international Pseudomonas aeruginosa consortium. Front. Microbiol. 2015, 6, 1036. [Google Scholar] [CrossRef] [Green Version]
- Römling, U.; Wingender, J.; Müller, H.; Tümmler, B. A major Pseudomonas aeruginosa clone common to patients and aquatic habitats. Appl. Environ. Microbiol. 1994, 60, 1734–1738. [Google Scholar] [CrossRef] [Green Version]
- Ozer, E.A.; Nnah, E.; Didelot, X.; Whitaker, R.J.; Hauser, A.R. The population structure of Pseudomonas aeruginosa is characterized by genetic isolation of exoU+ and exoS+ lineages. Genome Biol. Evol. 2019, 11, 1780–1796. [Google Scholar] [CrossRef] [Green Version]
- Freschi, L.; Vincent, A.T.; Jeukens, J.; Emond-Rheault, J.G.; Kukavica-Ibrulj, I.; Dupont, M.-J.; Charette, S.J.; Boyle, B.; Levesque, R.C. The Pseudomonas aeruginosa pan-genome provides new insights on its population structure, horizontal gene transfer, and pathogenicity. Genome Biol. Evol. 2019, 11, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Rahim, R.; Ochsner, U.; Olvera, C.; Graninger, M.; Messner, P.; Lam, J.S.; Soberón-Chávez, G. Cloning and functional characterization of the Pseudomonas aeruginosa rhlC gene that encodes rhamnosyltransferase 2, an enzyme responsible for di-rhamnolipid biosynthesis. Mol. Microbiol. 2001, 40, 708–718. [Google Scholar] [CrossRef]
- Mavrodi, D.V.; Bonsall, R.F.; Delaney, S.M.; Soule, M.J.; Phillips, G.; Thomashow, L.S. Functional analysis of genes for biosynthesis of pyocyanin and phenazine-1-carboxamide from Pseudomonas aeruginosa PAO1. J. Bacteriol. 2001, 183, 6454–6465. [Google Scholar] [CrossRef] [Green Version]
- Winsor, G.L.; Griffiths, E.J.; Lo, R.; Dhillon, B.K.; Shay, J.A.; Brinkman, F.S. Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res. 2016, 44, D646–D653. [Google Scholar] [CrossRef] [Green Version]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darling, A.E.; Mau, B.; Perna, N.T. ProgressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2- approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Baldini, R.L.; Déziel, E.; Saucier, M.; Zhang, Q.; Liberati, N.T.; Lee, D.; Urbach, J.; Goodman, H.M.; Rahme, L.G. The broad host range pathogen Pseudomonas aeruginosa strain PA14 carries two pathogenicity islands harboring plant and animal virulence genes. Proc. Natl. Acad. Sci. USA 2004, 101, 2530–2535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltrus, D.A.; Feng, Q.; Kvitko, B.H. Genome context influences evolutionary flexibility of nearly identical type III effectors in two phytopathogenic Pseudomonads. Front. Microbiol. 2022, 13, 826365. [Google Scholar] [CrossRef]
- Harrison, E.M.; Carter, M.E.; Luck, S.; Ou, H.Y.; He, X.; Deng, Z.; O’Callaghan, C.; Kadioglu, A.; Rajakumar, K. Pathogenicity islands PAPI-1 and PAPI-2 contribute individually and synergistically to the virulence of Pseudomonas aeruginosa strain PA14. Infect. Immun. 2010, 78, 1437–1446. [Google Scholar] [CrossRef] [Green Version]
- Foulkes, D.M.; McLean, K.; Haneef, A.S.; Ferning, D.G.; Winstanley, C.; Berry, N.; Kaye, S.B. Pseudomonas aeruginosa toxin ExoU as a therapeutic target in the treatment of bacterial infections. Microorganisms 2019, 7, 707. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Gurkar, A.U.; Lory, S. Interstrain transfer of the large pathogenicity island (PAPI-1) of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2006, 103, 19830–19835. [Google Scholar] [CrossRef] [Green Version]
- Turnbull, L.; Toyofuku, M.; Hynen, A.L.; Kurosawa, M.; Pessi, G.; Petty, N.K.; Osvath, S.R.; Cárcamo-Oyarce, G.; Gloag, E.S.; Shimoni, R.; et al. Explosive cell lysis as a mechanism for the biogenesis of bacterial membrane vesicles and biofilms. Nat. Commun. 2016, 7, 11220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel-Briand, Y.; Baysse, C. The pyocins of Pseudomonas aeruginosa. Biochimie 2002, 84, 499–510. [Google Scholar] [CrossRef]
- Nolan, L.M.; Turnbull, L.; Katrib, M.; Osvath, S.R.; Losa, D.; Lazenby, J.J.; Whitchurch, C.B. Pseudomonas aeruginosa is capable of natural transformation in biofilms. Microbiol.-SGM 2020, 166, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Nomura, N.; Suzuki, S. Biofilms: Hot spots of horizontal gene transfer (HGT) in aquatic environments, with a focus on a new HGT mechanism. FEMS Microbiol. Ecol. 2020, 96, fiaa031. [Google Scholar] [CrossRef] [PubMed]
- Reboud, E.; Elsen, S.; Bouillot, S.; Golovkine, G.; Basso, P.; Jeannot, K.; Attrée, I.; Huber, P. Phenotype and toxicity of the recently discovered exlA-positive Pseudomonas aeruginosa strains collected worldwide. Environ. Microbiol. 2016, 18, 3425–3439. [Google Scholar] [CrossRef] [PubMed]
- Trouillon, J.; Sentausa, E.; Ragno, M.; Robert-Genthon, M.; Lory, S.; Attrée, I.; Elsen, S. Species-specific recruitment of transcription factors dictates toxin expression. Nucleic Acids Res. 2020, 48, 2388–2400. [Google Scholar] [CrossRef] [Green Version]
- Berry, A.; Han, K.; Trouillon, J.; Robert-Genthon, M.; Ragno, M.; Lory, S.; Attrée, I.; Elsen, S. cAMP and Vfr control exolysin expression and cytotoxicity of Pseudomonas aeruginosa taxonomic outliers. J. Bacteriol. 2018, 200, e00135-18. [Google Scholar] [CrossRef] [Green Version]
- García-Reyes, S.; Moustafa, D.A.; Attrée, I.; Goldberg, J.B.; Quiroz-Morales, S.E.; Soberón-Chávez, G. Vfr or CyaB promote the expression of the pore-forming toxin exlBA operon in Pseudomonas aeruginosa ATCC 9027 without increasing its virulence in mice. Microbiol.-SGM 2021, 167, 001083. [Google Scholar] [CrossRef]
- García-Reyes, S.; Soto-Aceves, M.P.; Cocotl-Yañez, M.; González-Valdez, A.; Servín-González, L.; Soberón-Chávez, G. The outlier Pseudomonas aeruginosa strain ATCC 9027 harbors a defective LasR quorum-sensing transcriptional regulator. FEMS Microbiol. Lett. 2020, 367, fnaa122. [Google Scholar] [CrossRef]
- García-Reyes, S.; Cocotl-Yañez, M.; Soto-Aceves, M.P.; González-Valdez, A.; Servín-González, L.; Soberón-Chávez, G. The PqsR-independent quorum-sensing response of Pseudomonas aeruginosa ATCC 9027 outlier-strain reveals new insights on the PqsE effect on RhlR activity. Mol. Microbiol. 2021, 116, 1113–1123. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Roldán, L.; Rojo-Bezares, B.; de Toro, M.; López, M.; Toledano, P.; Lozano, C.; Chichón, G.; Alvarez-Erviti, L.; Torres, C.; Sáenz, Y. Antimicrobial resistance and virulence of Pseudomonas spp. among healthy animals: Concern about exolysin ExlA detection. Sci. Rep. 2020, 10, 11667. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Carranza, E.; García-Reyes, S.; González-Valdez, A.; Soberón-Chávez, G. Tracking the genome of four Pseudomonas aeruginosa isolates that have a defective Las quorum-sensing system but are still virulent. Access Microbiol. 2020, 2, acmi000132. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, E.L.; Vicente, A.C.P. Commentary: Clinical utilization of genomics data produced by the international Pseudomonas aeruginosa consortium. Front. Microbiol. 2016, 7, 770. [Google Scholar] [CrossRef] [PubMed]
- García-Ulloa, M.; Ponce-Soto, G.Y.; González-Valdez, A.; González-Pedrajo, B.; Díaz-Guerrero, M.; Souza, V.; Soberón-Chávez, G. Two Pseudomonas aeruginosa phylogroups belonging to the PA14 clade are indigenous to the Churince system in Cuatro Ciénegas Coahuila, México. Environ. Microbiol. 2019, 21, 2964–2976. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| STRAIN | PA14_51550 (PAPI-2) | PA14-14300 | Source of Isolation | Place of Isolation | Accession Number |
|---|---|---|---|---|---|
| WH-SGI-V-07678 | + | − | Hospital | United States | GCF_001453545.1 |
| AZPAE14698 | + | − | Respiratory tract infection | Israel | GCF_000794705.1 |
| AZPAE15028 | + | − | Respiratory tract infection | France | GCF_000792265.1 |
| AZPAE15028 | + | − | Unknown | United States | GCF_007021965.1 |
| PA99 | + | − | Urine | United States | GCF_000611995.2 |
| AUS122 | + | − | River, environmental | Australia | GCF_003976545.1 |
| T4242 | + | − | Sputum | Thailand | GCF_003975125.1 |
| MRSN1739 | + | − | Blood | United States | GCF_003969695.1 |
| ENV-208 | + | + | Residual water, environmental | Estonia | GCF_003633235.1 |
| VET-77 | + | − | Dog ear secretion | Estonia | GCF_003631375.1 |
| VET-39-D2 | + | − | Dog ear secretion | Estonia | GCF_003631365.1 |
| VET-44 | + | − | Ear secretion | Estonia | GCF_003631355.1 |
| PABL031 | + | − | Blood | United States | GCF_003411975.1 |
| L25 | + | − | Hospital effluent | Brazil | GCF_003402335.1 |
| M12 | + | − | Hospital effluent | Brazil | GCF_003402275.1 |
| WCHPA075056 | + | − | Clinical isolate | China | GCF_002976215.1 |
| WCHPA075063 | + | − | Clinical isolate | China | GCF_002976195.1 |
| CCF_716 | + | − | Clinical isolate | N/A | GCF_001910215.1 |
| WH-SGI-V-07638 | + | − | Hospital | United States | GCF_001453435.1 |
| AZPAE12423 | + | − | Cystic Fibrosis | Cleveland, United States | GCF_000797355.1 |
| AZPAE12418 | + | − | Cystic Fibrosis | Cleveland, United States | GCF_000797245.1 |
| AZPAE12416 | + | − | Cystic Fibrosis | Cleveland, United States | GCF_000797185.1 |
| AZPAE14899 | + | − | Urinary tract infection | Chennai, India | GCF_000791065.1 |
| ATCC 25324 | Incomplete genome | + | Glass shredder, air, environmental | Unknown | GCF_000297295.1 |
| 6093 | + | − | Cystic Fibrosis, sputum | Quebec, Canada | GCF_004371325.1 |
| MRSN16344 | + | − | Wound | United States | GCF_003969715.1 |
| AUS476 | + | − | Cystic Fibrosis | Australia | GCF_003840865.1 |
| CN573=PSE143 | + | − | Pleural liquid | Georgia | GCF_003836905.1 |
| A1(A2448) | + | − | Urine | Thailand | GCF_003835445.1 |
| HUM-257 | + | − | Bronchus-alveolar wash | Estonia | GCF_003631435.1 |
| M28A1 | + | − | Cow manure | Colombia | GCF_002117025.1 |
| BK5 | + | − | Eye, keratitis | Madurai, India | GCF_002242915.1 |
| isolate 406 | + | + | Unknown | EMBL | GCF_900147325.1 |
| isolate 405 | + | + | Unknown | EMBL | GCF_900147315.1 |
| 1046_PAER | + | − | Clinical isolate | United States | GCF_001060185.1 |
| AZPAE14960 | + | + | Respiratory tract infection | Spain | GCF_000791765.1 |
| 1163 | + | − | Blood | Brazil | GCF_009857765.1 |
| PA126 | + | − | Eye, keratitis | Australia | GCF_009727515.1 |
| AZPAE15052 | + | − | Respiratory tract | Argentina | GCF_000791465.1 |
| AZPAE14930 | Fragment | + | Urinary tract infection | Germany | GCF_000793865.1 |
| HM293 | Fragment | − | Bladder cancer | United Kingdom | GCF_003835395.1 |
| VET-35 | Fragment | + | Dog ear secretion | Estonia | GCF_003630095.1 |
| S700_C14_RS | Fragment | − | Respiratory tract infection | Italy | GCF_002135765.1 |
| PABL081 | Fragment | − | Blood | United States | GCF_003411045.1 |
| PABL075 | Fragment | + | Blood | United States | GCF_003410855.1 |
| PABL086 | Fragment | − | Blood | United States | GCF_003410925.1 |
| S708_C14_RS | Fragment | − | Respiratory tract infection | Italy | GCF_002136415.1 |
| PABL074 | Fragment | − | Blood | United States | GCF_003411025.1 |
| PA176 | Fragment | − | Eye | Australia | GCF_009727405.1 |
| STRAINS | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (1) PAO1 | 100 | |||||||||||||||
| (2) LESB65 | 99.35 | 100 | ||||||||||||||
| (3) PAK | 99.20 | 99.15 | 100 | |||||||||||||
| (4) WH-SGI-V-07678 | 99.29 | 99.15 | 99.07 | 100 | ||||||||||||
| (5) PA14 | 98.73 | 98.63 | 98.62 | 98.66 | 100 | |||||||||||
| (6) ID4365 | 98.71 | 98.62 | 98.54 | 98.59 | 98.96 | 100 | ||||||||||
| (7) PA_ W32 | 98.68 | 98.50 | 98.59 | 98.55 | 98.95 | 98.83 | 100 | |||||||||
| (8) AZPA E15016 | 98.69 | 98.56 | 98.58 | 98.57 | 99.95 | 98.93 | 98.85 | 100 | ||||||||
| (9) PA7 | 93.93 | 93.91 | 93.86 | 93.88 | 93.90 | 94.01 | 93.84 | 93.88 | 100 | |||||||
| (10) ATCC 9027 | 93.80 | 93.73 | 93.67 | 93.69 | 93.75 | 93.85 | 93.81 | 93.74 | 98.95 | 100 | ||||||
| (11) CLJ1 | 93.81 | 93.76 | 93.64 | 93.71 | 93.72 | 93.65 | 93.83 | 93.71 | 98.74 | 99.21 | 100 | |||||
| (12) LMG 5031 | 93.80 | 93.73 | 93.72 | 93.71 | 93.75 | 93.80 | 93.84 | 93.72 | 98.88 | 99.24 | 99.11 | 100 | ||||
| (13) PA-W1 | 97.46 | 97.44 | 97.35 | 97.40 | 97.28 | 97.35 | 97.34 | 97.24 | 93.80 | 93.70 | 93.74 | 93.71 | 100 | |||
| (14) CF_ PA39 | 97.50 | 97.47 | 97.36 | 97.44 | 97.29 | 97.34 | 97.30 | 97.26 | 93.84 | 93.75 | 93.77 | 93.71 | 99.50 | 100 | ||
| (15) 014-2A | 97.44 | 97.43 | 97.35 | 97.34 | 97.27 | 97.30 | 97.27 | 97.21 | 93.79 | 93.72 | 93.67 | 93.74 | 99.96 | 99.49 | 100 | |
| (16) ENV-567 | 97.45 | 97.38 | 97.34 | 97.37 | 97.26 | 97.31 | 97.28 | 97.16 | 93.80 | 93.67 | 93.69 | 93.69 | 99.49 | 99.49 | 99.44 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quiroz-Morales, S.E.; García-Reyes, S.; Ponce-Soto, G.Y.; Servín-González, L.; Soberón-Chávez, G. Tracking the Origins of Pseudomonas aeruginosa Phylogroups by Diversity and Evolutionary Analysis of Important Pathogenic Marker Genes. Diversity 2022, 14, 345. https://doi.org/10.3390/d14050345
Quiroz-Morales SE, García-Reyes S, Ponce-Soto GY, Servín-González L, Soberón-Chávez G. Tracking the Origins of Pseudomonas aeruginosa Phylogroups by Diversity and Evolutionary Analysis of Important Pathogenic Marker Genes. Diversity. 2022; 14(5):345. https://doi.org/10.3390/d14050345
Chicago/Turabian StyleQuiroz-Morales, Sara E., Selene García-Reyes, Gabriel Yaxal Ponce-Soto, Luis Servín-González, and Gloria Soberón-Chávez. 2022. "Tracking the Origins of Pseudomonas aeruginosa Phylogroups by Diversity and Evolutionary Analysis of Important Pathogenic Marker Genes" Diversity 14, no. 5: 345. https://doi.org/10.3390/d14050345
APA StyleQuiroz-Morales, S. E., García-Reyes, S., Ponce-Soto, G. Y., Servín-González, L., & Soberón-Chávez, G. (2022). Tracking the Origins of Pseudomonas aeruginosa Phylogroups by Diversity and Evolutionary Analysis of Important Pathogenic Marker Genes. Diversity, 14(5), 345. https://doi.org/10.3390/d14050345