
Patterns of Cryptic Diversity and Phylogeography in Four Freshwater Copepod Crustaceans in European Lakes


Abstract
1. Introduction
2. Materials and Methods
2.1. Overview of the Studied Taxa
2.2. Sampling
2.3. Molecular Analysis
2.4. Analysis of Sequences and Reconstruction of Phylogeny, Haplowebs and Population Structure
2.5. Species Delimitation
3. Results
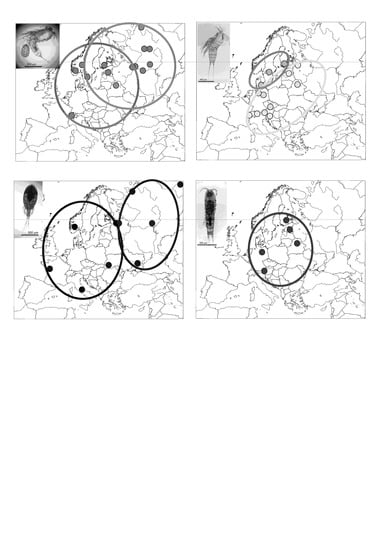
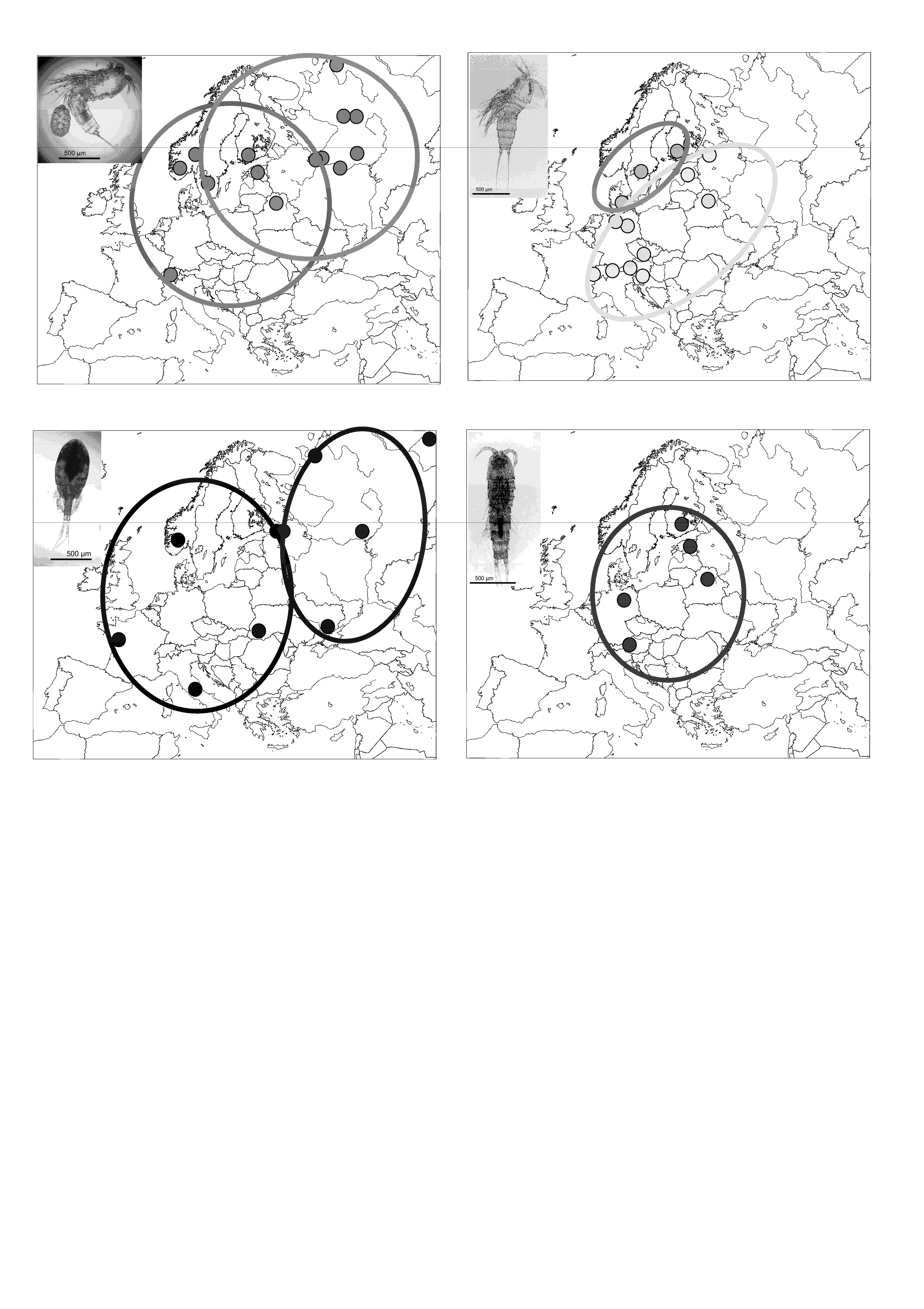
3.1. Attheyella crassa
3.2. Canthocamptus staphylinus
3.3. Eucyclops serrulatus
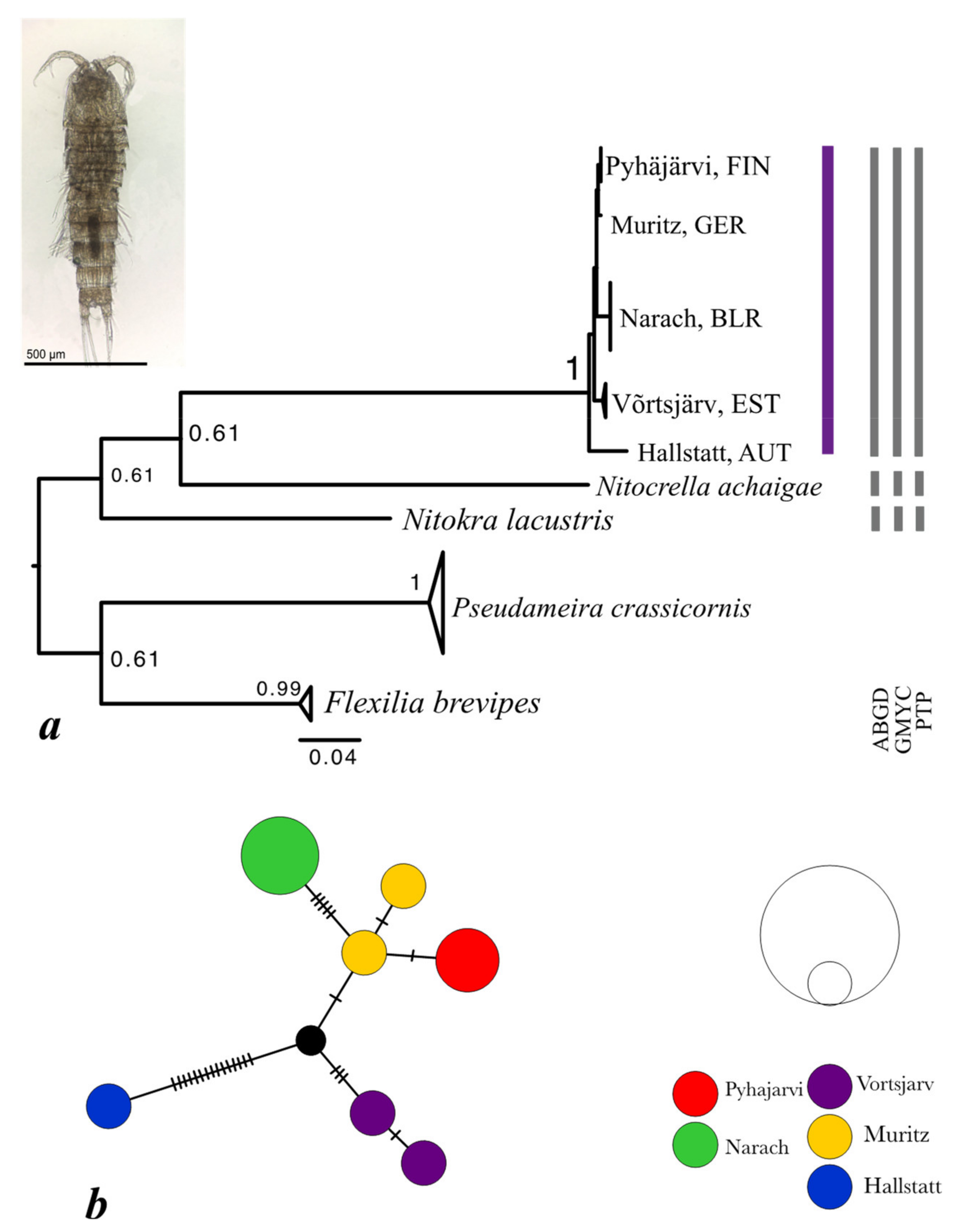
3.4. Nitokra hibernica
4. Discussion
4.1. Cryptic Diversification
4.2. Comparative Phylogeography
4.3. Demographic History
4.4. Age of Diversity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Incagnone, G.; Marrone, F.; Barone, R.; Robba, L.; Naselli-Flores, L. How do freshwater organisms cross the “dry ocean”? A review on passive dispersal and colonization processes with a special focus on temporary ponds. Hydrobiologia 2015, 750, 103–123. [Google Scholar] [CrossRef]
- Sudasinghe, H.; Pethiyagoda, R.; Ranasinghe, R.H.T.; Raghavan, R.; Dahanukar, N.; Meegaskumbura, M. A molecular phylogeny of the freshwater-fish genus Rasbora (Teleostei: Cyprinidae) in Sri Lanka reveals a remarkable diversification-And a cryptic species. J. Zool. Syst. Evol. Res. 2020, 58, 1076–1110. [Google Scholar] [CrossRef]
- Cordellier, M.; Pfenninger, M. Inferring the past to predict the future: Climate modelling predictions and phylogeography for the freshwater gastropod Radix balthica (Pulmonata, Basommatophora). Mol. Ecol. 2009, 18, 534–544. [Google Scholar] [CrossRef]
- Bekker, E.I.; Karabanov, D.P.; Galimov, Y.R.; Haag, C.R.; Neretina, T.V.; Kotov, A.A. Phylogeography of Daphnia magna Straus (Crustacea: Cladocera) in Northern Eurasia: Evidence for a deep longitudinal split between mitochondrial lineages. PLoS ONE 2018, 13, e0194045. [Google Scholar] [CrossRef]
- Neretina, A.N.; Karabanov, D.P.; Sacherova, V.; Kotov, A.A. Unexpected mitochondrial lineage diversity within the genus Alonella Sars, 1862 (Crustacea: Cladocera) across the Northern Hemisphere. PeerJ 2021, 9, e10804. [Google Scholar] [CrossRef]
- Zuykova, E.I.; Bochkarev, N.A.; Taylor, D.J.; Kotov, A.A. Unexpected endemism in the Daphnia longispina complex (Crustacea: Cladocera) in Southern Siberia. PLoS ONE 2019, 14, e0221527. [Google Scholar] [CrossRef] [PubMed]
- Verovnik, R.; Sket, B.; Trontelj, P. The colonization of Europe by the freshwater crustacean Asellus aquaticus (Crustacea: Isopoda) proceeded from ancient refugia and was directed by habitat connectivity. Mol. Ecol. 2005, 14, 4355–4369. [Google Scholar] [CrossRef]
- Vainio, J.K.; Väinölä, R. Refugial races and postglacial colonization history of the freshwater amphipod Gammarus lacustris in Northern Europe. Biol. J. Linn. Soc. 2003, 79, 523–542. [Google Scholar] [CrossRef]
- Weiss, M.; Macher, J.N.; Seefeldt, M.A.; Leese, F. Molecular evidence for further overlooked species within the Gammarus fossarum complex (Crustacea: Amphipoda). Hydrobiologia 2014, 721, 165–184. [Google Scholar] [CrossRef]
- Westram, A.M.; Jokela, J.; Keller, I. Hidden biodiversity in an ecologically important freshwater amphipod: Differences in genetic structure between two cryptic species. PLoS ONE 2013, 8, e69576. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Olivares, A.; Fleeger, J.W.; Foltz, D.W. Decoupling of molecular and morphological evolution in deep lineages of a meiobenthic harpacticoid copepod. Mol. Biol. Evol. 2001, 18, 1088–1102. [Google Scholar] [CrossRef]
- Pfenninger, M.; Schwenk, K. Cryptic animal species are homogeneously distributed among taxa and biogeographical regions. BMC Evol. Biol. 2007, 7, 121. [Google Scholar] [CrossRef]
- García-Morales, A.E.; Elías-Gutiérrez, M. DNA barcoding of freshwater Rotifera in Mexico: Evidence of cryptic speciation in common rotifers. Mol. Ecol. Resour. 2013, 13, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Ventura, M.; Vidal, N.; Gim, J.-S.; Buchaca, T.; Barmuta, L.A.; Jeppesen, E.; Joo, G.-J. Discovering hidden biodiversity: The use of complementary monitoring of fish diet based on DNA barcoding in freshwater ecosystems. Ecol. Evol. 2016, 6, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Macher, J.N.; Salis, R.K.; Blakemore, K.S.; Tollrian, R.; Matthaei, C.D.; Leese, F. Multiple-stressor effects on stream invertebrates: DNA barcoding reveals contrasting responses of cryptic mayfly species. Ecol. Indic. 2016, 61, 159–169. [Google Scholar] [CrossRef]
- Pereira, L.H.G.; Pazian, M.F.; Hanner, R.; Foresti, F.; Oliveira, C. DNA barcoding reveals hidden diversity in the Neotropical freshwater fish Piabina argentea (Characiformes: Characidae) from the Upper Paran Basin of Brazil. Mitochondrial DNA 2011, 22 (Suppl. 1), 87–96. [Google Scholar] [CrossRef]
- Fujisawa, T.; Barraclough, T.G. Delimiting Species Using Single-Locus Data and the Generalized Mixed Yule Coalescent Approach: A Revised Method and Evaluation on Simulated Data Sets. Syst. Biol. 2013, 62, 707–724. [Google Scholar] [CrossRef]
- Kapli, P.; Lutteropp, S.; Zhang, J.; Kobert, K.; Pavlidis, P.; Stamatakis, A.; Flouri, T. Phylogenetics Multi-rate Poisson tree processes for single-locus species delimitation under maximum likelihood and Markov chain Monte Carlo. Bioinformatics 2017, 33, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Puillandre, N.; Lambert, A.; Brouillet, S.; Achaz, G. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol. Ecol. 2012, 21, 1864–1877. [Google Scholar] [CrossRef]
- Cornils, A.; Held, C. Evidence of cryptic and pseudocryptic speciation in the Paracalanus parvus species complex (Crustacea, Copepoda, Calanoida). Front. Zool. 2014, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Previšić, A.; Gelemanović, A.; Urbanič, G.; Ternjej, I. Cryptic diversity in the Western Balkan endemic copepod: Four species in one? Mol. Phylogenet. Evol. 2016, 100, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Rossel, S.; Martínez Arbizu, P. Revealing higher than expected diversity of Harpacticoida (Crustacea:Copepoda) in the North Sea using MALDI-TOF MS and molecular barcoding. Sci. Rep. 2019, 9, 9182. [Google Scholar] [CrossRef] [PubMed]
- Sworobowicz, L.; Grabowski, M.; Mamos, T.; Burzyński, A.; Kilikowska, A.; Sell, J.; Wysocka, A. Revisiting the phylogeography of Asellus aquaticus in Europe: Insights into cryptic diversity and spatiotemporal diversification. Freshw. Biol. 2015, 60, 1824–1840. [Google Scholar] [CrossRef]
- Vakati, V.; Eyun, S.I.; Lee, W. Unraveling the intricate biodiversity of the benthic harpacticoid genus Nannopus (Copepoda, Harpacticoida, Nannopodidae) in Korean waters. Molecular Phylogenetics Evol. 2019, 130, 366–379. [Google Scholar] [CrossRef] [PubMed]
- Hamrová, E.; Krajicek, M.; Karanovic, T.; Černý, M.; Petrusek, A. Congruent patterns of lineage diversity in two species complexes of planktonic crustaceans, Daphnia longispina (Cladocera) and Eucyclops serrulatus (Copepoda), in East European mountain lakes. Zool. J. Linn. Soc. 2012, 166, 754–767. [Google Scholar] [CrossRef]
- Marrone, F.; Lo Brutto, S.; Hundsdoerfer, A.K.; Arculeo, M. Overlooked cryptic endemism in copepods: Systematics and natural history of the calanoid subgenus Occidodiaptomus Borutzky 1991 (Copepoda, Calanoida, Diaptomidae). Mol. Phylogenet. Evol. 2013, 66, 190–202. [Google Scholar] [CrossRef]
- Laakkonen, H.M.; Hardman, M.; Strelkov, P.; Väinölä, R. Cycles of trans-Arctic dispersal and vicariance, and diversification of the amphi-boreal marine fauna. J. Evol. Biol. 2021, 34, 73–96. [Google Scholar] [CrossRef]
- Avise, J.C.; Bowen, B.W.; Ayala, F.J. In the light of evolution X: Comparative phylogeography. Proc. Natl. Acad. Sci. USA 2016, 113, 7957–7961. [Google Scholar] [CrossRef]
- Wallace, A.R. The Geographical Distribution of Animals; With a Study of the Relations of Living and Extinct Faunas as Elucidating the Past Changes of the Earth’s Surface; Macmillan & Co.: London, UK, 1876; Volume 2. [Google Scholar]
- Riddle, B.R. Comparative phylogeography clarifies the complexity and problems of continental distribution that drove A. R. Wallace to favor islands. Proc. Natl. Acad. Sci. USA 2016, 113, 7970–7977. [Google Scholar] [CrossRef]
- Wood, D.A.; Vandergast, A.G.; Barr, K.R.; Inman, R.D.; Esque, T.C.; Nussear, K.E.; Fisher, R.N. Comparative phylogeography reveals deep lineages and regional evolutionary hotspots in the Mojave and Sonoran Deserts. Divers. Distrib. 2013, 19, 722–737. [Google Scholar] [CrossRef]
- Marko, P.B. “What’s larvae got to do with it?” Disparate patterns of post-glacial population structure in two benthic marine gastropods with identical dispersal potential. Mol. Ecol. 2004, 13, 597–611. [Google Scholar] [CrossRef]
- Hewitt, G.M. Genetic consequences of climatic oscillations in the Quaternary. Philos. Trans. R. Soc. B Biol. Sci. 2004, 359, 183–195. [Google Scholar] [CrossRef]
- Babik, W.; Branicki, W.; Crnobrnja-Isailović, J.; Cogǎlniceanu, D.; Sas, I.; Olgun, K.; Poyarkov, N.A.; Garcia-París, M.; Arntzen, J.W. Phylogeography of two European newt species—Discordance between mtDNA and morphology. Mol. Ecol. 2005, 14, 2475–2491. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M.; Santucci, F.; Reeve, N.J.; Hewitt, G.M. DNA footprints of European hedgehogs, Erinaceus europaeus and E. concolor: Pleistocene refugia, postglacial expansion and colonization routes. Mol. Ecol. 2001, 10, 2187–2198. [Google Scholar] [CrossRef] [PubMed]
- Nascetti, G.; Zangari, F.; Canestrelli, D. The spectacled salamanders, Salamandrina terdigitata (Lacépède, 1788) and S. perspicillata (Savi, 1821): Genetic differentiation and evolutionary history. Rend. Lincei 2005, 16, 159–169. [Google Scholar] [CrossRef]
- Paulo, O.S.; Dias, C.; Bruford, M.W.; Jordan, W.C.; Nichols, R.A. The persistence of Pliocene populations through the Pleistocene climatic cycles: Evidence from the phylogeography of an Iberian lizard. Proceedings of the Royal Society B. Biol. Sci. 2001, 268, 1625–1630. [Google Scholar] [CrossRef]
- Ursenbacher, S.; Schweiger, S.; Tomović, L.; Crnobrnja-Isailović, J.; Fumagalli, L.; Mayer, W. Molecular phylogeography of the nose-horned viper (Vipera ammodytes, Linnaeus (1758)): Evidence for high genetic diversity and multiple refugia in the Balkan peninsula. Mol. Phylogenet. Evol. 2008, 46, 1116–1128. [Google Scholar] [CrossRef]
- Ho, S. The Molecular clock and estimating species divergence. Nat. Educ. 2008, 1, 168. [Google Scholar]
- Jażdżewska, A.M.; Rewicz, T.; Mamos, T.; Wattier, R.; Bącela-Spychalska, K.; Grabowski, M. Cryptic diversity and mtDNA phylogeography of the invasive demon shrimp, Dikerogammarus haemobaphes (Eichwald, 1841), in Europe. NeoBiota 2020, 57, 53–86. [Google Scholar] [CrossRef]
- Bron, J.E.; Frisch, D.; Goetze, E.; Johnson, S.C.; Lee, C.E.; Wyngaard, G.A. Observing copepods through a genomic lens. Front. Zool. 2011, 8, 22. [Google Scholar] [CrossRef]
- Regier, J.C.; Shultz, J.W.; Kambic, R.E. Pancrustacean phylogeny: Hexapods are terrestrial crustaceans and maxillopods are not monophyletic. Proc. R. Soc. B Biol. Sci. 2005, 272, 395–401. [Google Scholar] [CrossRef]
- Borutzky, E.V. Freshwater Harpacticoida. Crustacea. Freshwater Harpacticoida. Fauna of the USSR; USSR Academy of Sciences: Moscow, Russia, 1952; Volume 3, 424p. (In Russian) [Google Scholar]
- Easton, E.E.; Thistle, D.; Spears, T. Species boundaries in Zausodes-complex species (Copepoda: Harpacticoida: Harpacticidae) from the north-eastern Gulf of Mexico. Invertebr. Syst. 2010, 24, 258–270. [Google Scholar] [CrossRef]
- Lee, C.E. Global phylogeography of a cryptic copepod species complex and reproductive isolation between genetically proximate “populations”. Evolution 2000, 54, 2014–2027. [Google Scholar] [CrossRef]
- Questel, J.M.; Blanco-Bercial, L.; Hopcroft, R.R.; Bucklin, A. Phylogeography and connectivity of the Pseudocalanus (Copepoda: Calanoida) species complex in the eastern North Pacific and the Pacific Arctic Region. J. Plankton Res. 2016, 38, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Winkler, G.; Souissi, S.; Poux, C.; Castric, V. Genetic heterogeneity among Eurytemora affinis populations in Western Europe. Mar. Biol. 2011, 158, 1841–1856. [Google Scholar] [CrossRef]
- Sukhikh, N.; Alekseev, V. Genetic and morphological heterogeneity within Eucyclops serrulatus (Fischer, 1851) (Crustacea: Copepoda: Cyclopidae). J. Nat. Hist. 2015, 49, 2929–2953. [Google Scholar] [CrossRef]
- Hołyńska, M.; Wyngaard, G.A. Towards a phylogeny of Cyclops (Copepoda): (in) Congruences among morphology, molecules and zoogeography. Zool. Scr. 2019, 48, 376–398. [Google Scholar] [CrossRef]
- Karanovic, T.; Cooper, S.J.B. Explosive radiation of the genus Schizopera on a small subterranean island in Western Australia (Copepoda:Harpacticoida): Unravelling the cases of cryptic speciation, size differentiation and multiple invasions. Invertebr. Syst. 2012, 26, 115–192. [Google Scholar] [CrossRef]
- Kochanova, E.S.; Gaviria, S. Integrative taxonomy of the freshwater harpacticoid Attheyella crassa G.O. Sars, 1863 (Crustacea: Copepoda: Canthocamptidae) in the Palearctic region. Invertebr. Zool. 2018, 15, 267–276. [Google Scholar] [CrossRef]
- Kochanova, E.; Sarvala, J.; Fefilova, E. Phylogenetic Study of Dioecious and Parthenogenetic Populations of Canthocamptus staphylinus (Crustacea, Copepoda, Harpacticoida). Bull. South. Calif. Acad. Sci. 2018, 117, 138–149. [Google Scholar] [CrossRef][Green Version]
- Sars, G.O. Oversigt af de indenlandske Ferskvandcopepoder. Forh. i Vidensk.-Selsk. i Christiania Aar 1862, 1863, 212–262. [Google Scholar]
- Jurine, L. Histoire Des Monocles, Qui Se Trouvent Aux Environs De Genève; Paschoud, J.J., Ed.; Genève et Paris: Paris, France, 1820; 258p. [Google Scholar]
- Fischer, S. Beitrage zur Kenntnis der in der Umgegend von St Petersburg sich findenden Cyclopiden. Bull. Société Impériale Nat. Moscou 1851, 24, 409–438. [Google Scholar]
- Brady, G.S. A Monograph of the Free and Semi-Parasitic Copepoda of the British Islands; Ray Society: London, UK, 1880; Volume 3, pp. 83–93. [Google Scholar]
- Enckell, P.H. Kräftdjur (Fältfauna); Signum: Lund, Sweden, 1980; 420p. [Google Scholar]
- Defaye, D.; Dussart, N. World Directory of Crustacea Copepoda of Inland Waters, II-Harpacticoides 2011; Editions N. Boubée & Cie: Paris, Frence; 450p.
- Gardeström, J.; Dahl, U.; Kotsalainen, O.; Maxson, A.; Elfwing, T.; Grahn, M.; Bengtsson, B.E.; Breitholtz, M. Evidence of population genetic effects of long-term exposure to contaminated sediments—A multi-endpoint study with copepods. Aquat. Toxicol. 2008, 86, 426–436. [Google Scholar] [CrossRef]
- Turesson, E.U.; Stiernström, S.; Minten, J.; Adolfsson-Erici, M.; Bengtsson, B.E.; Breitholtz, M. Development and reproduction of the freshwater harpacticoid copepod Attheyella crassa for assessing sediment-associated toxicity. Aquat. Toxicol. 2007, 83, 180–189. [Google Scholar] [CrossRef]
- Lang, K. Monographie der Harpacticiden; Håkan Ohlsson: Lund, German, 1948; 1682p. [Google Scholar]
- Sarvala, J. A parthenogenetic life cycle in a population of Canthocamptus staphylinus (Copepoda, Harpacticoida). Hydrobiologia 1979, 62, 113–129. [Google Scholar] [CrossRef]
- Smyly, W.J.P. Observations on the life-history of the harpacticoid copepod, Canthocamptus staphylinus (Jurine). Ann. Mag. Nat. Hist. 1957, 10, 509–512. [Google Scholar] [CrossRef]
- Alekseev, V.R.; Defaye, D. Taxonomic differentiation and world geographical distribution of the Eucyclops serrulatus group (Copepoda, Cyclopidae, Eucyclopinae). In Studies on Freshwater Copepoda: A Volume in Honour of Bernard Dussart. Crustaceana Monographs; Defaye, D., Suárez, E., von Vaupel Klein, J.C., Alekseev, V., Eds.; Brill: Leiden, The Netherlands, 2011; Volume 16, pp. 41–72. [Google Scholar]
- Grigorovich, I.A.; Dovgal, I.V.; MacIsaac, H.J.; Monchenko, V.I. Acineta nitocrae: A new suctorian epizootic on nonindigenous harpacticoid copepods, Nitocra hibernica and N. incerta, in the Laurentian Great Lakes. Arch. Hydrobiol. 2001, 152, 161–176. [Google Scholar] [CrossRef]
- Lesko, L.T.; Hudson, P.L.; Reid, J.W.; Chriscinske, M.A. Harpacticoid Copepods of the Laurentian Great Lakes; Great Lakes Science Center: Ann Arbor, MI, USA, 2003. Available online: http://www.glsc.usgs.gov/greatlakescopepods/Key.asp?GROUP=Harpacticoid (accessed on 13 September 2021).
- Duggan, I.C.; Van Overdijk, C.D.A.; Bailey, S.A.; Jenkins, P.T.; Limén, H.; MacIsaac, H.J. Invertebrates associated with residual ballast water and sediments of cargo-carrying ships entering the Great Lakes. Can. J. Fish. Aquat. Sci. 2005, 62, 2463–2474. [Google Scholar] [CrossRef]
- Hudson, P.L.; Reid, J.W.; Lesko, L.T.; Selgeby, J.H. Cyclopoid and harpacticoid copepods of the Laurentian Great Lakes. Ohio Biol. Surv. Bull. New Ser. 1998, 12, 1–50. [Google Scholar]
- Aljanabi, S.M.; Martinez, I. Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nucleic Acids Res. 1997, 25, 4692–4693. [Google Scholar] [CrossRef] [PubMed]
- Walsh, P.S.; Metzger, D.A.; Higushi, R. Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 1991, 10, 506–513. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; Mcgettigan, P.A.; Mcwilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinform. Appl. Note 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Posada, D. jModelTest: Phylogenetic model averaging. Molecular Biology and Evolution. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP*. Phylogenetic Analysis Using Parsimony (* and Other Methods); Version 4; Sinauer Associates: Sunderland, UK, 2003. [Google Scholar]
- Young, S.-S.; Lee, Y.-Y.; Liu, M.-Y. Genetic variability and divergence of Neutrodiaptomus tumidus Kiefer 1937 (Copepoda: Calonida) among 10 subpopulations in the high mountain range of Taiwan and their phylogeographical relationships indicated by mtDNA COI gene. Zool. Stud. 2014, 53, 22. [Google Scholar] [CrossRef]
- Thum, R.A.; Harrison, R.G. Deep genetic divergences among morphologically similar and parapatric Skistodiaptomus (Copepoda: Calanoida: Diaptomidae) challenge the hypothesis of Pleistocene speciation. Biol. J. Linn. Soc. 2009, 96, 150–165. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef]
- Pons, J.; Barraclough, T.G.; Gomez-Zurita, J.; Cardoso, A.; Duran, D.P.; Hazell, S.; Kamoun, S.; Sumlin, W.D.; Vogler, A.P. Sequence-Based Species Delimitation for the DNA Taxonomy of Undescribed Insects. Syst. Biol. 2006, 55, 595–609. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kü Hnert, D.; Vaughan, T.; Wu, C.-H. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed]
- Jones, G. Mathematical Biology Algorithmic improvements to species delimitation and phylogeny estimation under the multispecies coalescent. J. Math. Biol. 2017, 74, 447–467. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Copyedited by: YS MANUSCRIPT CATEGORY: Software for Systematics and Evolution Software for Systematics and Evolution Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 2. [Google Scholar] [CrossRef]
- Burton, R.S.; Byrne, R.J.; Rawson, P.D. Three divergent mitochondrial genomes from California populations of the copepod Tigriopus californicus. Gene 2007, 403, 53–59. [Google Scholar] [CrossRef]
- Garlitska, L.; Neretina, T.; Schepetov, D.; Mugue, N.; De Troch, M.; Baguley, J.G.; Azovsky, A. Cryptic diversity of the “cosmopolitan” harpacticoid copepod Nannopus palustris: Genetic and morphological evidence. Mol. Ecol. 2012, 21, 5336–5347. [Google Scholar] [CrossRef]
- Willett, C.S.; Ladner, J.T. Investigations of fine-scale phylogeography in Tigriopus californicus reveal historical patterns of population divergence. BMC Evol. Biol. 2009, 9, 139. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.O.; Ueda, H. Genetic analysis on Pseudodiaptomus inopinus (Copepoda, Calanoida) species complex in Japan: Revival of the species name of P. japonicus Kikuchi, 1928. Plankton Benthos Res. 2018, 13, 173–179. [Google Scholar] [CrossRef]
- Barreto, F.S.; Watson, E.T.; Lima, T.G.; Willett, C.S.; Edmands, S.; Li, W.; Burton, R.S. Genomic signatures of mitonuclear coevolution across populations of Tigriopus californicus. Nat. Ecol. Evol. 2018, 2, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, N.J.; Figueroa, D.F.; Hicks, D. Phylogeography of Acartia tonsa Dana, 1849 (Calanoida: Copepoda) and phylogenetic reconstruction of the genus Acartia Dana, 1846. Mar. Biodivers. 2020, 50, 23. [Google Scholar] [CrossRef]
- Brancelj, A.; Karanovic, T. A new subterranean Maraenobiotus (Crustacea: Copepoda) from Slovenia challenges the concept of polymorphic and widely distributed harpacticoids. J. Nat. Hist. 2015, 49, 2905–2928. [Google Scholar] [CrossRef]
- Cox, A.J.; Hebert, P.D.N. Colonization, extinction, and phylogeographic patterning in a freshwater crustaceanfile: Evolutionary genomics in the Metazoa-the mitochondrial DNA as a model system. Mol. Ecol. 2001, 10, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Huemer, P.; Hebert, P.D.N.; Mutanen, M.; Wieser, C.; Wiesmair, B.; Hausmann, A.; Yakovlev, R.; Möst, M.; Gottsberger, B.; Strutzenberger, P.; et al. Large geographic distance versus small DNA barcode divergence: Insights from a comparison of European to South Siberian Lepidoptera. PLoS ONE 2018, 13, e0206668. [Google Scholar] [CrossRef] [PubMed]
- Pentinsaari, M.; Hebert, P.D.N.; Mutanen, M. Barcoding beetles: A regional survey of 1872 species reveals high identification success and unusually deep interspecific divergences. PLoS ONE 2014, 9, e108651. [Google Scholar] [CrossRef]
- Rawson, P.D.; Burton, R.S. Molecular evolution at the cytochrome oxidase subunit 2 gene among divergent populations of the intertidal copepod, Tigriopus californicus. J. Mol. Evol. 2006, 62, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Ketmaier, V.; Marrone, F.; Alfonso, G.; Paulus, K.; Wiemann, A.; Tiedemann, R.; Mura, G. Mitochondrial DNA regionalism and historical demography in the extant populations of Chirocephalus kerkyrensis (Branchiopoda: Anostraca). PLoS ONE 2012, 7, e30082. [Google Scholar] [CrossRef]
- Knowlton, N.; Weigt, L.A. New dates and new rates for divergence across the Isthmus of Panama. Proc. R. Soc. B Biol. Sci. 1998, 265, 2257–2263. [Google Scholar] [CrossRef]
- Marino, I.A.M.; Pujolar, J.M.; Zane, L. Reconciling deep calibration and demographic history: Bayesian inference of post glacial colonization patterns in Carcinus aestuarii (Nardo, 1847) and C. maenas (Linnaeus, 1758). PLoS ONE 2011, 6, e28567. [Google Scholar] [CrossRef]
- Zofkova, M.; Timms, B.V. A conflict of morphological and genetic patterns in the australian anostracan Branchinella longirostris. Hydrobiologia 2009, 635, 67–80. [Google Scholar] [CrossRef]
- Sworobowicz, L.; Mamos, T.; Grabowski, M.; Wysocka, A. Lasting through the ice age: The role of the proglacial refugia in the maintenance of genetic diversity, population growth, and high dispersal rate in a widespread freshwater crustacean. Freshw. Biol. 2020, 65, 1028–1046. [Google Scholar] [CrossRef]
- Burton, R.S.; Pereira, R.J.; Barreto, F.S. Cytonuclear genomic interactions and hybrid breakdown. Annu. Rev. Ecol. Evol. Syst. 2013, 44, 281–302. [Google Scholar] [CrossRef]
- Barreto, F.S.; Moy, G.W.; Burton, R.S. Interpopulation patterns of divergence and selection across the transcriptome of the copepod Tigriopus californicus. Mol. Ecol. 2011, 20, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Goetze, E. Cryptic speciation on the high seas; global phylogenetics of the copepod family Eucalanidae. Proc. R. Soc. B Biol. Sci. 2003, 270, 2321–2331. [Google Scholar] [CrossRef]
- Lee, C.E. Rapid and repeated invasions of fresh water by the copepod Eurytemora affinis. Evolution 1999, 53, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.E.; Frost, B.W. Morphological stasis in the Eurytemora affinis species complex (Copepoda: Temoridae). Hydrobiologia 2002, 480, 102–115. [Google Scholar] [CrossRef]
- Soltis, D.E.; Morris, A.B.; McLachlan, J.S.; Manos, P.S.; Soltis, P.S. Comparative phylogeography of unglaciated eastern North America. Mol. Ecol. 2006, 15, 4261–4293. [Google Scholar] [CrossRef]
- Bowen, B.W.; Gaither, M.R.; DiBattista, J.D.; Iacchei, M.; Andrews, K.R.; Grant, W.S.; Toonen, R.J.; Briggs, J.C. Comparative phylogeography of the ocean planet. Proc. Natl. Acad. Sci. USA 2016, 113, 7962–7969. [Google Scholar] [CrossRef]
- Bickford, D.; Lohman, D.J.; Sodhi, N.S.; Ng, P.K.L.; Meier, R.; Winker, K.; Ingram, K.K.; Das, I. Cryptic species as a window on diversity and conservation. Trends Ecol. Evol. 2007, 22, 148–155. [Google Scholar] [CrossRef]
- Dawson, M.N. Parallel phylogeographic structure in ecologically similar sympatric sister taxa. Mol. Ecol. 2012, 21, 987–1004. [Google Scholar] [CrossRef]
- Kreger, K.M.; Shaban, B.; Wapstra, E.; Burridge, C.P. Phylogeographic parallelism: Concordant patterns in closely related species illuminate underlying mechanisms in the historically glaciated Tasmanian landscape. J. Biogeogr. 2020, 47, 1674–1686. [Google Scholar] [CrossRef]
- Siivonen, L. The history of the Fennoscandian mammal fauna. Acta Zool. Fenn. 1982, 169, 7–10. [Google Scholar]
- Buchovska, J.; Danusevičius, D.; Baniulis, D.; Stanys, V.; Šikšnianiene, J.B.; Kavaliauskas, D. The location of the Northern glacial refugium of scots pine based on mitochondrial DNA markers. Balt. For. 2013, 19, 2–12. [Google Scholar]
- Dering, M.; Kosiński, P.; Wyka, T.P.; Pers-Kamczyc, E.; Boratyński, A.; Boratyńska, K.; Reich, P.B.; Romo, A.; Zadworny, M.; Żytkowiak, R.; et al. Tertiary remnants and Holocene colonizers: Genetic structure and phylogeography of Scots pine reveal higher genetic diversity in young boreal than in relict Mediterranean populations and a dual colonization of Fennoscandia. Divers. Distrib. 2017, 23, 540–555. [Google Scholar] [CrossRef]
- Jensen, T.C.; Walseng, B.; Hessen, D.O.; Dimante-Deimantovica, I.; Novichkova, A.A.; Chertoprud, E.S.; Chertoprud, M.V.; Sakharova, E.G.; Krylov, A.V.; Frisch, D.; et al. Changes in trophic state and aquatic communities in high Arctic ponds in response to increasing goose populations. Freshw. Biol. 2019, 64, 1241–1254. [Google Scholar] [CrossRef]
- Najdenski, H.; Dimova, T.; Zaharieva, M.M.; Nikolov, B.; Petrova-Dinkova, G.; Dalakchieva, S.; Popov, K.; Tokarevich, N.K. Migratory birds along the Mediterranean—Black Sea Flyway as carriers of zoonotic pathogens. Can. J. Microbiol. 2018, 64, 915–924. [Google Scholar] [CrossRef]
- Lee, C.E. Evolutionary mechanisms of habitat invasions, using the copepod Eurytemora affinis as a model system. Evol. Appl. 2016, 9, 248–270. [Google Scholar] [CrossRef]
- Hartl, D.L.; Clark, A.G. Principles of Population Genetics, 4th ed.; Sinauer Associates: Franklin County, MA, USA, 2007. [Google Scholar]
- Pichler, F.B. Genetic Assessment of Population Boundries and Gene Exchange in Hector’s Dolphin; Department of Conservation: Wellington, New Zealand, 2002; Volume 44, pp. 1–37. [Google Scholar]
- Gollner, S.; Stuckas, H.; Kihara, T.C.; Laurent, S.; Kodami, S.; Arbizu, P.M. Mitochondrial DNA analyses indicate high diversity, expansive population growth and high genetic connectivity of vent copepods (Dirivultidae) across different oceans. PLoS ONE 2016, 11, e0163776. [Google Scholar] [CrossRef]
- Gerlach, S.A. On the importance of marine meiofauna for benthos communities. Oecologia 1971, 6, 176–190. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | N of Sequences | Aligned Sequence Length (bp) | π | Hd | N of Haplotypes | S | Eta | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mtDNA | rRNA | mtDNA | rRNA | mtDNA | rRNA | mtDNA | rRNA | mtDNA | rRNA | mtDNA | rRNA | mtDNA | rRNA | |
| Attheyella crassa | 37 | 17 | 545 | 461 | 0.093 | 0.033 | 0.868 | 0.831 | 12 | 6 | 77 | 32 | 81 | 32 |
| Canthocamptus staphylinus | 59 | 10 | 606 | 1576 | 0.103 | 0.001 | 0.94 | 0.64 | 30 | 4 | 179 | 3 | 205 | 3 |
| Eucyclops serrulatus | 38 | 12 | 654 | 628 | 0.193 | 0.011 | 0.862 | 0.758 | 12 | 5 | 131 | 11 | 157 | 11 |
| Nitokra hibernica | 10 | - | 554 | - | 0.013 | - | 0.911 | - | 7 | - | 26 | - | 26 | - |
| Species/Clade | Western Clade | Eastern Clade | A. nordenskioldii | A. dentata |
|---|---|---|---|---|
| W clade | 1.3/0.26 | 6.48 | - | - |
| E clade | 26.1 | 5.5/0.62 | - | - |
| A. nordenskioldii | 34.5 | 32.4 | - | - |
| A. dentata | 35.8 | 42.5 | 36.1 | 0.8/- |
| Species | Clade | Population | N | (%) | Hd/h | Tajima’s D | Fu’s Fs |
|---|---|---|---|---|---|---|---|
| A. crassa | E clade | Pääjärvi | 3 | 1.1 | 1.0/3 | - | - |
| Hiiu | 1 | - | -/1 | - | - | ||
| Vänern | 2 | 0.18 | 1.0/2 | - | - | ||
| Nyuvchim | 3 | 0.0 | 0.0/1 | - | - | ||
| Pechora | 1 | - | -/1 | - | - | ||
| Karkalai | 2 | 0.9 | 1.0/2 | - | - | ||
| Narach | 4 | 0.09 | 0.5/2 | −0.61 | 0.172 | ||
| Vychegda | 1 | - | -/1 | - | - | ||
| Rybinsk | 3 | 0 | 0/1 | - | - | ||
| Glubokoe | 1 | - | -/1 | - | - | ||
| Total clade | 21 | 0.78 | 0.91/15 | - | - | ||
| W clade | Geneva | 3 | 0. 1 | 0.66/2 | - | - | |
| Sognsvann | 6 | 0.07 | 0.33/2 | −0.93 | −0.003 | ||
| Rybinsk | 7 | 0 | 0/1 | - | - | ||
| Total clade | 16 | 3.2 | 0.61/5 | - | - | ||
| C. staphylinus | F clade | Pääjärvi | 11 | 0 | 0/1 | - | - |
| Vänern | 9 | 0.7 | 0.97/8 | −0.74 | −2.91 * | ||
| Total clade | 21 | 0.015 | 0.73/9 | - | - | ||
| C clade | Geneva | 8 | 0.5 | 0.64/4 | −1.79 * | 1.34 | |
| Zurich | 1 | - | -/1 | - | - | ||
| Võrtsjärv | 5 | 0.0 | 0.0/1 | - | - | ||
| Narach | 5 | 1.0 | 0.9/5 | 1.61 | 1.28 | ||
| Orlov | 10 | 0.9 | 0.64/5 | 0.24 | 3.56 * | ||
| Plau am see | 2 | 0.0 | 0.0/1 | - | - | ||
| Stechlin | 2 | 0.0 | 0.0/1 | - | - | ||
| Fernsteinsee | 1 | - | -/1 | - | - | ||
| Zell am see | 2 | 0.0 | 0.0/1 | - | - | ||
| Hallstatt | 1 | - | -/1 | - | - | ||
| Total clade | 37 | 2.2 | 0.94/21 | ||||
| E. serrulatus | E clade | SevDvina | 1 | - | -/1 | - | - |
| Lena | 1 | - | -/1 | - | - | ||
| Karkalai | 6 | 0.9 | 0.33/2 | −1.33 | 2.59 | ||
| Dniester | 5 | 0.0 | 0.0/1 | - | - | ||
| Orlov | 2 | 1.7 | 1/2 | - | - | ||
| Tauride | 1 | - | -/1 | - | - | ||
| Total clade | 15 | 1.0 | 0.79/8 | - | - | ||
| W clade | Oslo | 3 | 0.7 | 0.66/2 | - | - | |
| Florence | 2 | 0.0 | 0.0/1 | - | - | ||
| Creteil | 2 | 0.0 | 0.0/1 | - | - | ||
| Transkarpathia | 5 | 0.0 | 0.0/1 | - | - | ||
| Orlov | 6 | 0.7 | 0.8/4 | 0.28 | 0.82 | ||
| Tauride | 3 | 1.1 | 0.66/2 | - | - | ||
| Total clade | 20 | 1.0 | 0.85/11 | - | - | ||
| N. hibernica | Narach | 3 | 0.0 | 0.0/3 | - | - | |
| Võrtsjärv | 2 | 0.18 | 2/1 | - | - | ||
| Pyhäjärvi | 2 | 0.0 | 0.0/1 | - | - | ||
| Hallstatt | 1 | - | - | - | - | ||
| Müritz | 2 | 0.18 | 2/1 | - | - | ||
| Total clade | 10 | 1.3 | 0.91/6 | - | - |
| Species/Clade | F Clade (Fennoscandian) | C Clade (Continental) | C. microstaphylinus | C. (B.) longifurcatus | C. kitaurensis |
|---|---|---|---|---|---|
| F clade | 2.8/0.003 | 0.007 | - | - | - |
| C clade | 33.2 | 3.6/0.007 | - | - | - |
| C. microstaphylinus | 38.2 | 43.3 | 1.1 | - | - |
| C. (B.) longifurcatus | 34.2 | 32.1 | 41.0 | - | - |
| C. kitaurensis | 46.4 | 15.3 | 44.6 | 39.9 | - |
| Species/clade | E clade | W clade | E. cuatrocienegas | E. prionophorus | E. taiwanensis |
|---|---|---|---|---|---|
| E clade | 2.1/0.34 | 2.41 | - | - | - |
| W clade | 36.6 | 1.3/0.22 | - | - | - |
| E. cuatrocienegas | 43.2 | 43.5 | - | - | - |
| E. prionophorus | 48.4 | 43.6 | 38.5 | 0.52/- | - |
| E. taiwanensis | 39.1 | 42.2 | 41.3 | 41.6 | 0.44/- |
| Species | N. hibernica | N. lacustris |
|---|---|---|
| Nitokra hibernica | 3.6 | - |
| Nitokra lacustris | 47.3 | - |
| Nitocrella achaiae | 53.4 | 49.7 |
| Species | Order | Model | Г Distribution Parameter (α) | Inter-Clade Distances, % | |||
|---|---|---|---|---|---|---|---|
| Model Distances GTR+Г | p-Distances | “Split Age” Ma | Data Reference | ||||
| Attheyella crassa | Harpacticoida | GTR+Г | 0.58 | 26.1 | 16.4 | 7—19 | [51]; this study |
| Canthocamptus staphylinus | Harpacticoida | GTR+Г | 0.50 | 33.2 | 17.4 | 8—23 | [52]; this study |
| Eucyclops serrulatus | Cyclopoida | GTR+Г | 0.59 | 36.6 | 20.6 | 9—26 | [48]; this study |
| Eudiaptomus hadzici | Calanoida | GTR+Г | 0.69 | 17.6–42.1 | 12.3–24.0 | [21] | |
| Neutrodiaptomus tumidus | Calanoida | GTR+Г | 0.19 | 15.2–28.3 | 6.7–9.4 | [76] | |
| Hemidiaptomus ingens | Calanoida | GTR+Г | 0.21 | 28.2–56.3 | 11.4–17.7 | [26] | |
| Skistodiaptomus pallidus | Calanoida | GTR+Г | 0.17 | 33.9–48.1 | 16.0–17.2 | [77] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kochanova, E.; Nair, A.; Sukhikh, N.; Väinölä, R.; Husby, A. Patterns of Cryptic Diversity and Phylogeography in Four Freshwater Copepod Crustaceans in European Lakes. Diversity 2021, 13, 448. https://doi.org/10.3390/d13090448
Kochanova E, Nair A, Sukhikh N, Väinölä R, Husby A. Patterns of Cryptic Diversity and Phylogeography in Four Freshwater Copepod Crustaceans in European Lakes. Diversity. 2021; 13(9):448. https://doi.org/10.3390/d13090448
Chicago/Turabian StyleKochanova, Elena, Abhilash Nair, Natalia Sukhikh, Risto Väinölä, and Arild Husby. 2021. "Patterns of Cryptic Diversity and Phylogeography in Four Freshwater Copepod Crustaceans in European Lakes" Diversity 13, no. 9: 448. https://doi.org/10.3390/d13090448
APA StyleKochanova, E., Nair, A., Sukhikh, N., Väinölä, R., & Husby, A. (2021). Patterns of Cryptic Diversity and Phylogeography in Four Freshwater Copepod Crustaceans in European Lakes. Diversity, 13(9), 448. https://doi.org/10.3390/d13090448