Abstract
This work presents an updated and more complete version of the transcriptome of a long-distance migrant, the Northern Wheatear (Oenanthe oenanthe). The improved transcriptome was produced from the independent mRNA sequencing of adipose tissue, brain, intestines, liver, skin, and muscle tissues sampled during the autumnal migratory season. This new transcriptome has better sequencing coverage and is more representative of the species’ migratory phenotype. We assembled 20,248 transcripts grouped into 16,430 genes, from which 78% were successfully annotated. All the standard assembly quality parameters were improved in the second transcriptome version.
1. Introduction
The transcriptome represents a very dynamic part of the cellular machinery. The gene-specific mRNA lifetime is strictly controlled because it is one of the first levels in the regulation of cellular and developmental processes [1,2]. Therefore, the transcriptome-level analysis of a tissue is a proxy to the biological functions it performs and to the phenotype of an organism [3,4].
Sequencing the transcriptome of non-model species is a challenge but is of special value. Species-specific sequence information can be used a priori for further functional studies and the understanding of specific phenotypes such as adaptations for migration in birds [5,6].
For molecular ecology and conservation studies, transcriptomic information can be used to infer population diversity and resilience [7,8,9,10] and not just mere genetic polymorphisms [11,12]. Furthermore, the novel assembly of transcriptomes opens the door for orthologous comparisons of biological functions among distinct species, with implications for discovering novel approaches to treat, e.g., metabolic diseases in humans [13,14].
Bird Migration
The study of the birds’ migratory phenotype is of special interest since it shows the unique conjunction of a healthy super-fat phenotype and a simultaneous increment of physical activity [15,16], rarely found among other vertebrates. Thus, migratory birds have evolved to efficiently manage lipid metabolism and to counteract obesity-related diseases [17,18,19,20,21].
RNA-Seq techniques are powerful tools for the study of complex traits and the assembly of a high quality reference transcriptome is a necessary first step to guarantee reliable results [22].
As a part of the integrative study of the migratory phenotype of the Northern Wheatear (Oenanthe oenanthe), we previously published a preliminary and partial transcriptome [23]. In this work, we present an improved version of that transcriptome, which was obtained by adding sequence information from the assembly of transcriptome analyses of six tissues: the brain, intestines, liver, adipose tissue, muscle, and skin. Both sets of RNA-Seq data were combined and analyzed together. This transcriptome is, therefore, representative of the set of active genes of this migrating species.
2. Materials and Methods
This paper is part of a series that analyses seasonal adaptations for migration of the Northern Wheatear (Oenanthe oenanthe) [6,24]. The sequence data analyzed in this manuscript derive from the same birds used in Frias-Soler et al. (2018) [23] and Frias-Soler et al. (2020) [6]. Therefore, here, we only briefly describe the procedures used to collect the samples and isolate the RNA.
The birds used in this study were bred at the Institute of Avian Research (Wilhelmshaven, Germany). The samples were collected from birds at three different stages of migratory fueling: lean birds, birds while fattening, and fat birds after reaching their body-mass plateau (three birds per body mass condition). Migratory fattening was induced by subjecting the birds to a constant light–dark cycle of 12 h light:12 h dark at 20 ± 1 °C room temperature following the end of the summer [25]. The samples were collected from the brain, intestines, liver, pectoral muscles, adipose tissue, and skin.
For RNA purification, a GeneMATRIX Universal RNA Purification Kit (Roboklon GmbH, Berlin, Germany) was used, and for the final clean-up, we used an RNeasy Fibrous Tissue Mini Kit (Qiagen GmbH, Hilden, Germany). We followed the suppliers’ protocols and the general recommendations for RNA isolation given by Sambrook et al. (1989) [26].
2.1. Sequencing and Transcriptome Assembly
RNA samples (28 s/18 s ratio ≥ 1.7; RIN ≥ 8; A260/A280 ≥ 1.8) from three individuals were pooled together by tissue (brain, intestines, liver, adipose tissue, muscle, or skin) and body mass conditions and sent to GATC-Biotech AG (Konstanz, Germany) for sequencing. Samples were sequenced using a Genome Sequencer Illumina HiSeq2000 device (Sequence mode: 50 bp single-end reads). GATC-Biotech conducted cDNA library preparation following the company’s standard protocols. From the total RNA sample poly(A)+ RNA was isolated and first-strand cDNA synthesis was primed with a N6 randomized primer. Then adapters were ligated to the 5′ and 3′ ends of the cDNA and it was finally amplified by PCR.
The reads were trimmed using Trimmomatic. v1 [27], homopolymers (60% over the entire length of the read represented by one nucleotide), primers, and adapters, and ambiguous residues (Ns) from both sides of the sequence were trimmed. The base pairs with a Phred-score of less than 20 were removed from the ends. After this step, reads with a length of <36 bases were discarded. Sam tools version 1.3.1 [28] was employed to generate the mapping statistics and filter out unmapped reads.
2.2. De Novo Assembly and Post-Processing
The short-reads from all tissues and conditions (n = 18) were pulled together to assemble de novo the transcriptome of O. oenanthe using Trinity, version 2.6.5 [29], which was run under the following conditions: in silico normalization of reads (normalize_reads and normalize_max_read_cov 50) and set for single-end reads fastq data input.
The new transcriptome was merged in a fasta file with the previous version [23] sequenced using the Genome Sequencer Roche GS FLX System generating 400 bp length reads. The redundant transcripts were then removed using CD-Hit-EST [30] by merging transcripts with a sequence similarity of 0.9. Low expressed transcripts that accounted for at most 10% of the overall expression (cumulative sum of reads by transcript) were discarded. In this way, we obtained a new, compact, and non-redundant transcriptome for O. oenanthe.
To estimate the completeness of the transcriptome, the conserved ortholog gene content was estimated using the Benchmarking Universal Single-Copy Orthologs (BUSCO) tool, version 3.0.2 [31]. The analysis was conducted against the available avian information in the database aves_odb9. The standard transcript quality-control length-parameters were also computed. To assess the quality and summarize the key standard parameters of transcriptome assemble, we used the TransRate tool [32].
To check the quality of the assembly, the reads were remapped by tissue to the new transcriptome using the mem-algorithm implemented in BWA-MEM version 0.7.12 [33] while flagging potential chimeras like supplementary reads.
2.3. Annotation
Transcripts were annotated to the coding sequences of Ficedula albicollis (release 96, downloaded from ftp://ftp.ensembl.org/pub/release-96/fasta/ficedula_albicollis/cds/; accessed in 15 May 2019) and the UniProtKB/Swiss-Prot 2019_05 databases using Blastn and Blastx, respectively. For the alignment, an E-value = 0.005 was set, and the best hit (maximum Bitscore) from any of the two sources of annotation was selected for naming the transcripts.
Based on the annotation, we summarized all transcripts with a unique “gene symbol” into a single O. oenanthe gene (OOENG). If one “Trinity gene” clustered multiple transcripts that were annotated to different genes, those genes were split into different “oenanthe genes” to avoid ambiguity. For unannotated transcripts, either the code derived from Trinity or the original isotig/isogroup from the first transcriptome version was kept.
The R programming language platform, version 3.2.2, was employed for statistical analysis and plotting [34].
The raw sequence data and the assembled transcriptome can be found in the Transcriptome Shotgun Assembly (TSA) database from the National Center for Biotechnology Information (NCBI; https://www.ncbi.nlm.nih.gov; accessed in 20 February 2021) under the accession number TSA GFYT00000000 version 2. The full annotated transcriptome can be found in the supplements (Table S1).
3. Results and Discussion
A more robust and complete transcriptome for O. oenanthe was obtained by combining the previously published data—one-time sequencing using ~400 bp long reads—with new transcripts derived from the assembly of short reads from the brain, intestines, liver, adipose tissue, muscle, and skin.
The percentages of the mapped reads by tissue to both transcriptomes are shown in Table 1. The new transcriptome recovered between 8 and 13% more reads than the original one. Meaning that if the old transcriptome were used for further differential gene expression analyses, these reads would remain unmapped with the consequent loss of information. Therefore, the new version is more representative of the “migratory transcriptome”, a fact that would increase the sensitivity and robustness of the future comparisons.

Table 1.
Percentage of reads and supplementary number of reads mapped to each tissue for both versions of the O. oenanthe transcriptome. Ten percent or more of the reads that did not map to any transcripts from the previous version are now mapped to the new transcriptome.
The new version contains 20,248 transcripts (the old version had 21,746); 13,040 (64%) are from the new assembled transcriptome, and 7208 (36%) are from the previous version. Up to 1112 new open reading frames (ORF) are described in the new transcriptome a good indicator of its improved quality [22]. Table 2 summarizes the parameters of both transcriptomes.
Table 2.
Transcriptome assembly statistics. Data generated by Transrate [32].
The new version has longer transcripts and over 47 million assembled bases (32% more than the previous one). Thus, the new version is potentially more complete and less fragmented than the old version (Figure 1).
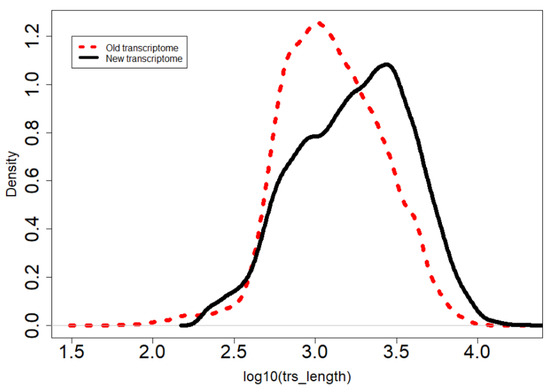
Figure 1.
The transcript length distribution of the new (solid line) and old (discontinuous line) transcriptomes. The new version has fewer and longer transcripts.
Transcriptome Completeness
The new O. oenanthe transcriptome shows better quality parameters regarding gene completeness (BUSCO scores) than the first version. It agrees with the previous findings of longer transcripts and higher number of reported ORFs (Table 3). The percentages of complete and single-copy complete genes refer to the orthologous near-universally-distributed avian genes found in the wheatears’ transcriptome [31]. The new version was enriched in approximately 30% of these genes, a reliable indicator of its higher quality.
Table 3.
The Benchmarking Universal Single-Copy Orthologs (BUSCO) scores from the new and old version of the O. oenanthe transcriptomes.
The 20,248 assembled transcripts were attributable to 16,430 genes, between the reported number of avian genes: 9909 in the Sunbird Asity (Neodrepanis coruscans) and 19,174 in Zebra Finch (Taeniopygia guttata) [35]. In total, 10,311 of these genes were fully annotated: 12,164 (60%) to F. albicollis, 2455 (12%) to the Swiss-Prot database, and 5631 (28%) unannotated. Up to 6596 isoforms were found which correspond to 2768 genes (Table 4). In the new version, as in the previous one, the gene with the highest number of isoforms is titin. These transcripts are most likely gene fragments and not real isoforms (OOENG09682 length interval: 469–9168 bp) since titin is the largest gene identified in vertebrates [36].
Table 4.
The number of isoforms per “Oenanthe gene” found in the newly assembled transcriptome.
Titin fragmentation and the still modest completeness percentage of the new transcriptome suggest that sequencing the transcriptome of more tissues, sampled under different metabolic conditions using more in-depth sequencing coverage/longer reads, is needed to better describe the wheatears transcriptome.
In summary, in this paper, we presented an updated version of the Northern Wheatear transcriptome, which we recommend for use in further RNA-Seq studies on the species.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/d13040151/s1, the full annotated transcriptome can be found in the supplements Table S1.
Author Contributions
R.C.F.-S. wrote the paper, designed the methodology, analyzed and interpreted the data. L.V.P. participated on the conceptualization of the manuscript, the methodology design and conducting the bioinformatics analysis. M.W. and F.B. participated on the conceptualization of the manuscript, funding acquisition, supervision, reviewing and editing the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Institute of Pharmacy and Molecular Biotechnology, Heidelberg University, Germany and the Institute of Avian Research, Wilhelmshaven, Germany. Roberto Frias was supported by a fellowship from the German Academic Exchange Service (DAAD).
Institutional Review Board Statement
The study was conducted according to the guidelines of the German Animal Welfare Act and approved by the Zweckverband Veterinäramt JadeWeser (42508-Te).
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw sequence data and the assembled transcriptome can be found in the Transcriptome Shotgun Assembly (TSA) database from the National Center for Biotech-nology Information (NCBI; https://www.ncbi.nlm.nih.gov; accessed in 20 February 2021) under the accession number TSA GFYT00000000 version 2.
Acknowledgments
We want to thank Hedwig Sauer-Gürth and Heidi Staudter for their support in the laboratory work. Thanks to Agnes Hotz-Wagenblatt from the Bioinformatics Group, Core Facility Genomics and Proteomics, German Cancer Research Center, Heidelberg University, for her advice on bioinformatic support.
Conflicts of Interest
Michael Wink is the editor-in-chief of the journal Diversity.
References
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell; Taylor & Francis Group: Abingdon, UK, 2007. [Google Scholar]
- Lewin, B.; Krebs, J.; Goldstein, E.; Kilpatrick, S.T.; Goldstein, E.S. Lewin’s GENES X; Jones & Bartlett Learning: Burlington, MA, USA, 2011. [Google Scholar]
- Nelson, D.L.; Cox, M.M.; Lehninger, A.L. Lehninger Principles of Biochemistry; W.H. Freeman and Company: New York, NY, USA, 2013. [Google Scholar]
- Edfors, F.; Danielsson, F.; Hallström, B.M.; Käll, L.; Lundberg, E.; Pontén, F.; Forsström, B.; Uhlén, M. Gene-specific correlation of RNA and protein levels in human cells and tissues. Mol. Syst. Biol. 2016, 12, 883. [Google Scholar] [CrossRef] [PubMed]
- Jax, E.; Wink, M.; Kraus, R.H. Avian transcriptomics: Opportunities and challenges. J. Ornithol. 2018, 159, 599–629. [Google Scholar] [CrossRef]
- Frias-Soler, R.C.; Pildaín, L.V.; Pârâu, L.G.; Wink, M.; Bairlein, F. Transcriptome signatures in the brain of a migratory songbird. Comp. Biochem. Physiol. Part D Genom. Proteom. 2020, 34, 100681. [Google Scholar] [CrossRef]
- Alvarez, M.; Schrey, A.W.; Richards, C.L. Ten years of transcriptomics in wild populations: What have we learned about their ecology and evolution? Mol. Ecol. 2015, 24, 710–725. [Google Scholar] [CrossRef]
- Akman, M.; Carlson, J.E.; Holsinger, K.E.; Latimer, A.M. Transcriptome sequencing reveals population differentiation in gene expression linked to functional traits and environmental gradients in the South. African shrub Protea repens. New Phytol. 2016, 210, 295–309. [Google Scholar] [CrossRef]
- Watson, H.; Videvall, E.; Andersson, M.N.; Isaksson, C. Transcriptome analysis of a wild bird reveals physiological responses to the urban environment. Sci. Rep. 2017, 7, 44180. [Google Scholar] [CrossRef]
- Hao, Y.; Xiong, Y.; Cheng, Y.; Song, G.; Jia, C.; Qu, Y.; Lei, F. Comparative transcriptomics of 3 high-altitude passerine birds and their low-altitude relatives. Proc. Nat. Acad. Sci. USA 2019, 116, 11851–11856. [Google Scholar] [CrossRef]
- Hartl, D.L.; Clark, A.G.S. Principles of Population Genetic, 3rd ed.; Sinauer Associates Inc.: Sunderland, MA, USA, 1997; p. 682. [Google Scholar]
- Allendorf, F.W.; Luikart, G.H.; Aitken, S.N. Conservation and the Genetics of Populations, 2nd ed.; Wiley-Blackwell: Oxford, UK, 2013; p. 624. [Google Scholar]
- Gerstein, M.B.; Rozowsky, J.; Yan, K.-K.; Wang, D.; Cheng, C.; Brown, J.B.; Davis, C.A.; Hillier, L.; Sisu, C.; Li, J.J.; et al. Comparative analysis of the transcriptome across distant species. Nature 2014, 512, 445–448. [Google Scholar] [CrossRef]
- Gaye, A.; Doumatey, A.P.; Davis, S.K.; Rotimi, C.N.; Gibbons, G.H. Whole-genome transcriptomic insights into protective molecular mechanisms in metabolically healthy obese African Americans. NPJ Genom. Med. 2018, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Guglielmo, C.G. Obese super athletes: Fat-fueled migration in birds and bats. J. Exp. Biol. 2018, 221, jeb165753. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, S.R.; Guglielmo, C.; Pierce, B.; Klaassen, M. Flying, fasting, and feeding in birds during migration: A nutritional and physiological ecology perspective. J. Avian Biol. 2004, 35, 377–393. [Google Scholar] [CrossRef]
- Guglielmo, C.G. Move that fatty acid: Fuel selection and transport in migratory birds and bats. Integr. Comp. Biol. 2010, 50, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.J.; McWilliams, S.R. Seasonal changes in composition of lipid storages in migratory birds: Causes and consequences. Condor 2005, 107, 269–279. [Google Scholar] [CrossRef]
- Smith, S.B.; McWilliams, S.R.; Guglielmo, C.G. Effect of diet composition on plasma metabolite profiles in a migratory songbird. Condor 2007, 109, 48–58. [Google Scholar] [CrossRef]
- Maggini, I.; Bairlein, F. Endogenous rhythms of seasonal migratory body mass changes and nocturnal restlessness in different populations of Northern Wheatear Oenanthe oenanthe. J. Biol. Rhythms 2010, 25, 268–276. [Google Scholar] [CrossRef]
- Maggini, I.; Bulte, M.; Bairlein, F. Endogenous control of fuelling in a migratory songbird. Sci. Nat. 2017, 104, 93. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-cabrero, D.; Cervera, A.; McPherson, A.; Szcze, W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.A. survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 1–19. [Google Scholar] [CrossRef]
- Frias-Soler, R.C.; Villarín-Pildaín, L.; Hotz-Wagenblatt, A.; Kolibius, J.; Bairlein, F.; Wink, M. De novo annotation of the transcriptome of the Northern Wheatear (Oenanthe oenanthe). Peer J. 2018, 6, e5860. [Google Scholar] [CrossRef]
- Bairlein, F.; Eikenaar, C.; Schmaljohann, H. Routes to genes: Unravelling the control of avian migration—An integrated approach using Northern Wheatear. J. Ornithol. 2015, 156, 3–14. [Google Scholar] [CrossRef]
- Bulte, M.; Bairlein, F. Endogenous control of migratory behavior in Alaskan northern wheatears Oenanthe oenanthe. J. Ornithol. 2013, 154, 567–570. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1989. [Google Scholar]
- Bolger, A.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. Genome analysis BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Smith-Unna, R.; Boursnell, C.; Patro, R.; Hibberd, J.M.; Kelly, S. TransRate: Reference-free quality assessment of de novo transcriptome assemblies. Genome Res. 2016, 26, 1134–1144. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997v2. [Google Scholar]
- R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; R-Core-Team: Vienna, Austria, 2020. [Google Scholar]
- Feng, S.; Stiller, J.; Deng, Y.; Armstrong, J.; Fang, Q.; Reeve, A.H.; Xie, D.; Chen, G.; Guo, C.; Faircloth, B.C.; et al. Dense sampling of bird diversity increases power of comparative genomics. Nature 2020, 587, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Kulke, M.; Leake, M.C.; Neagoe, C.; Hinssen, H.; Hajjar, R.J.; Linke, W.A. Damped elastic recoil of the titin spring in myofibrils of human myocardium. Proc. Nat. Acad. Sci. USA 2003, 100, 12688–12693. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).