Mitochondrial Genomes from New Zealand’s Extinct Adzebills (Aves: Aptornithidae: Aptornis) Support a Sister-Taxon Relationship with the Afro-Madagascan Sarothruridae

 , , , , ,
, , , , ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Samples
2.2. DNA Extraction
2.2.1. Bone Samples
2.2.2. Tissue Samples
2.2.3. Eggshell Samples
2.3. Library Preparation and DNA Sequencing
2.3.1. Bone and Tissue Samples
2.3.2. Eggshell Samples
2.4. Data Processing
2.4.1. Bone and Tissue Samples
2.4.2. Eggshell Samples
2.5. Genome Assembly
2.6. Taxon Sampling and Sequence Alignment
2.7. Molecular Phylogenetic Analyses
2.8. Morphological Phylogenetic Analyses
2.9. Fossil Calibrations and Molecular Clock Analyses
3. Results
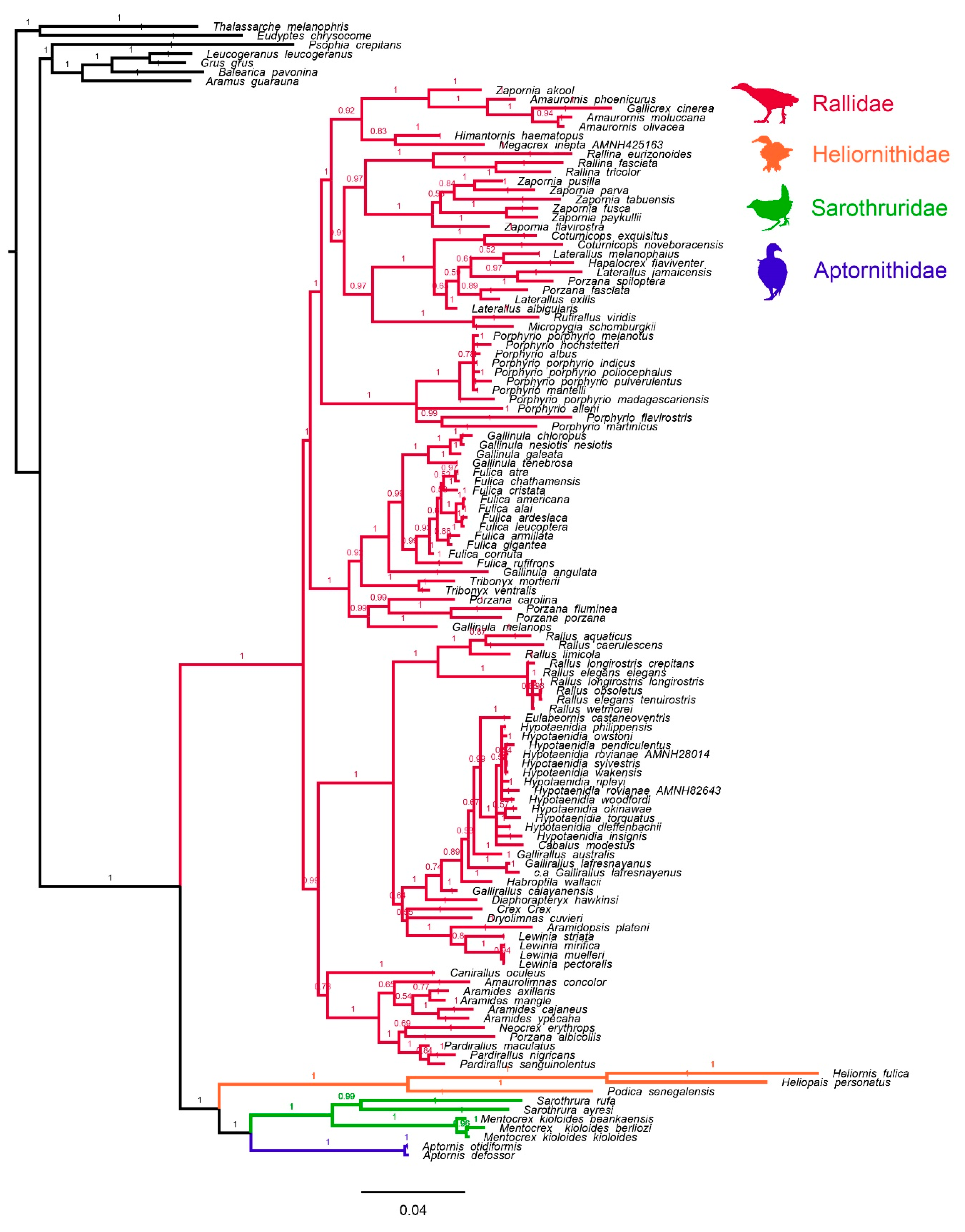
3.1. Mitochondrial Tree Topology
3.2. Morphological Character Evolution
3.3. Node Age Estimates
4. Discussion
Data Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schellart, W.P.; Lister, G.S.; Toy, V.G. A Late Cretaceous and Cenozoic reconstruction of the Southwest Pacific region: Tectonics controlled by subduction and slab rollback processes. Earth-Sci. Rev. 2006, 76, 191–233. [Google Scholar] [CrossRef]
- Worthy, T.H.; Holdaway, R.N. The Lost World of the MOA: Prehistoric Life of New Zealand; Indiana University Press: Bloomington, IN, USA, 2002; ISBN 0-253-34034-9. [Google Scholar]
- Tennyson, A.J.D.; Martinson, P. Extinct Birds of New Zealand; Te Papa Press: Wellington, New Zealand, 2006; ISBN 0-909010-21-8.
- Ericson, P.G.; Christidis, L.; Cooper, A.; Irestedt, M.; Jackson, J.; Johansson, U.S.; Norman, J.A. A Gondwanan origin of passerine birds supported by DNA sequences of the endemic New Zealand wrens. Proc. R. Soc. Lond. B Biol. Sci. 2002, 269, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Wright, T.F.; Schirtzinger, E.E.; Matsumoto, T.; Eberhard, J.R.; Graves, G.R.; Sanchez, J.J.; Capelli, S.; Müller, H.; Scharpegge, J.; Chambers, G.K. A multilocus molecular phylogeny of the parrots (Psittaciformes): Support for a Gondwanan origin during the Cretaceous. Mol. Biol. Evol. 2008, 25, 2141–2156. [Google Scholar] [CrossRef] [PubMed]
- Bunce, M.; Szulkin, M.; Lerner, H.R.; Barnes, I.; Shapiro, B.; Cooper, A.; Holdaway, R.N. Ancient DNA provides new insights into the evolutionary history of New Zealand’s extinct giant eagle. PLoS Biol. 2005, 3, e9. [Google Scholar] [CrossRef] [PubMed]
- Holdaway, R.N.; Worthy, T.H.; Tennyson, A.J. A working list of breeding bird species of the New Zealand region at first human contact. N. Z. J. Zool. 2001, 28, 119–187. [Google Scholar] [CrossRef]
- Wilmshurst, J.M.; Anderson, A.J.; Higham, T.F.G.; Worthy, T.H. Dating the late prehistoric dispersal of Polynesians to New Zealand using the commensal Pacific rat. Proc. Natl. Acad. Sci. USA 2008, 105, 7676–7680. [Google Scholar] [CrossRef] [PubMed]
- Cole, T.L.; Wood, J.R. The ancient DNA revolution: The latest era in unearthing New Zealand’s faunal history. N. Z. J. Zool. 2018, 45, 91–120. [Google Scholar] [CrossRef]
- Owen, R. On Dinornis, an Extinct Genus of Tridactyle Struthious Birds, with Descriptions of Portions of the Skeleton of Five Species which Formerly Existed in New Zealand (Part I). Trans. Zool. Soc. Lond. 1844, 3, 235–275, pls 18–30. [Google Scholar]
- Owen, R. On Dinornis (part III): Containing a description of the skull and beak of that genus, and of the same characteristic parts of Palapteryx, and of two other genera of birds, Notornis and Nestor; forming part of an extensive series of ornithic remains discovered by Mr Walter Mantell at Waingongoro, North Island of New Zealand. Trans. Zool. Soc. Lond. 1848, 3, 345–378. [Google Scholar]
- Owen, R. On Dinornis (part XV): Containing a description of the skull, femur, tibia, fibula, and metatarsus of Aptornis defossor, Owen, from near Oamaru, Middle Island, New Zealand; with additional observations on Aptornis otidiformis, on Notornis mantelli, and on Dinornis curtus. Trans. Zool. Soc. Lond. 1871, 7, 353–380. [Google Scholar]
- Mantell, G.A. On the fossil remains of birds collected in various parts of New Zealand by Mr. Walter Mantell, of Wellington. Q. J. Geol. Soc. 1848, 4, 225–238. [Google Scholar] [CrossRef]
- Worthy, T.H.; Tennyson, A.J.; Scofield, R.P. Fossils reveal an early Miocene presence of the aberrant gruiform Aves: Aptornithidae in New Zealand. J. Ornithol. 2011, 152, 669–680. [Google Scholar] [CrossRef]
- Wood, J.R.; Scofield, R.P.; Hamel, J.; Lalas, C.; Wilmshurst, J.M. Bone stable isotopes indicate a high trophic position for New Zealand’s extinct South Island adzebill (Aptornis defossor)(Gruiformes: Aptornithidae). N. Z. J. Ecol. 2017, 41, 240–244. [Google Scholar] [CrossRef]
- Trotter, M.M. Archaeological investigations in the Aviemore area, South Island. Rec. Canterb. Mus. 1970, 8, 439–453. [Google Scholar]
- Scofield, P.; Worthy, T.; Schlumpf, H. What birds were New Zealand’s first people eating? Wairau Bar’s avian remains re-examined. Rec. Canterb. Mus. 2003, 17, 17–35. [Google Scholar]
- Seersholm, F.V.; Cole, T.L.; Grealy, A.; Rawlence, N.J.; Greig, K.; Knapp, M.; Stat, M.; Hansen, A.J.; Easton, L.J.; Shepherd, L. Subsistence practices, past biodiversity, and anthropogenic impacts revealed by New Zealand-wide ancient DNA survey. Proc. Natl. Acad. Sci. USA 2018, 115, 7771–7776. [Google Scholar] [CrossRef] [PubMed]
- McWethy, D.B.; Whitlock, C.; Wilmshurst, J.M.; McGlone, M.S.; Fromont, M.; Li, X.; Dieffenbacher-Krall, A.; Hobbs, W.O.; Fritz, S.C.; Cook, E.R. Rapid landscape transformation in South Island, New Zealand, following initial Polynesian settlement. Proc. Natl. Acad. Sci. USA 2010, 107, 21343–21348. [Google Scholar] [CrossRef]
- Perry, G.L.W.; Wheeler, A.B.; Wood, J.R.; Wilmshurst, J.M. A high-precision chronology for the rapid extinction of New Zealand moa (Aves, Dinornithiformes). Quat. Sci. Rev. 2014, 105, 126–135. [Google Scholar] [CrossRef]
- Cracraft, J. Phylogenetic relationships and transantarctic biogeography of some gruiform birds. Geobios 1982, 15, 393–402. [Google Scholar] [CrossRef]
- Livezey, B.C. A phylogenetic analysis of the Gruiformes (Aves) based on morphological characters, with an emphasis on the rails (Rallidae). Philos. Trans. R. Soc. Lond. B Biol. Sci. 1998, 353, 2077–2151. [Google Scholar] [CrossRef]
- Livezey, B.C.; Zusi, R.L. Higher-order phylogeny of modern birds (Theropoda, Aves: Neornithes) based on comparative anatomy. II. Analysis and discussion. Zool. J. Linn. Soc. 2007, 149, 1–95. [Google Scholar] [CrossRef] [PubMed]
- Claramunt, S.; Cracraft, J. A new time tree reveals Earth history’s imprint on the evolution of modern birds. Sci. Adv. 2015, 1, e1501005. [Google Scholar] [CrossRef]
- Campbell, H.; Hutching, G. In Search of Ancient New Zealand; Penguin Books NZ: Auckland, New Zealand, 2007. [Google Scholar]
- Mortimer, N.; Campbell, H.J.; Tulloch, A.J.; King, P.R.; Stagpoole, V.M.; Wood, R.A.; Rattenbury, M.S.; Sutherland, R.; Adams, C.J.; Collot, J. Zealandia: Earth’s hidden continent. GSA Today 2017, 27, 27–35. [Google Scholar] [CrossRef]
- Fain, M.G.; Krajewski, C.; Houde, P. Phylogeny of “core Gruiformes” (Aves: Grues) and resolution of the Limpkin–Sungrebe problem. Mol. Phylogenet. Evol. 2007, 43, 515–529. [Google Scholar] [CrossRef] [PubMed]
- Hackett, S.J.; Kimball, R.T.; Reddy, S.; Bowie, R.C.; Braun, E.L.; Braun, M.J.; Chojnowski, J.L.; Cox, W.A.; Han, K.-L.; Harshman, J. A phylogenomic study of birds reveals their evolutionary history. Science 2008, 320, 1763–1768. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, E.D.; Mirarab, S.; Aberer, A.J.; Li, B.; Houde, P.; Li, C.; Ho, S.Y.; Faircloth, B.C.; Nabholz, B.; Howard, J.T. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 2014, 346, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Prum, R.O.; Berv, J.S.; Dornburg, A.; Field, D.J.; Townsend, J.P.; Lemmon, E.M.; Lemmon, A.R. A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Nature 2015, 526, 569–573. [Google Scholar] [CrossRef]
- Houde, P.; Cooper, A.; Leslie, E.; Strand, A.; Montano, G. Phylogeny and evolution of 12S rDNA in Gruiformes (Aves). Avian Mol. Evol. Syst. 1997, 121–158. [Google Scholar]
- Dickinson, E.C.; Remsen, J.V.J. The Howard and Moore Complete Checklist of the Birds of the World, 4th ed.; Volume 1 Non-Passerines; Aves Press: Eastbourne, UK, 2013. [Google Scholar]
- García-R, J.C.; Gibb, G.C.; Trewick, S.A. Deep global evolutionary radiation in birds: Diversification and trait evolution in the cosmopolitan bird family Rallidae. Mol. Phylogenet. Evol. 2014, 81, 96–108. [Google Scholar] [CrossRef]
- Worthy, T.H. Fossil Distribution of Brown Teal (Anas chlorotis) in New Zealand; DoC Science Internal Series 81; Department of Conservation: Wellington, UK, 2002; ISBN 0-478-22315-3.
- Worthy, T.; Holdaway, R.N. Quaternary fossil faunas from caves in Takaka Valley and on Takaka Hill, northwest Nelson, South Island, New Zealand. J. R. Soc. N. Z. 1994, 24, 297–391. [Google Scholar] [CrossRef]
- Worthy, T.H. Fossils of Honeycomb Hill; Museum of New Zealand: Wellington, New Zealand, 1993; ISBN 0-908953-04-6.
- Holdaway, R.N.; Roberts, R.G.; Beavan-Athfield, N.R.; Olley, J.M.; Worthy, T.H. Optical dating of quartz sediments and accelerator mass spectrometry 14C dating of bone gelatin and moa eggshell: A comparison of age estimates for non-archaeological deposits in New Zealand. J. R. Soc. N. Z. 2002, 32, 463–505. [Google Scholar] [CrossRef]
- Brotherton, P.; Haak, W.; Templeton, J.; Brandt, G.; Soubrier, J.; Adler, C.J.; Richards, S.M.; Der Sarkissian, C.; Ganslmeier, R.; Friederich, S. Neolithic mitochondrial haplogroup H genomes and the genetic origins of Europeans. Nat. Commun. 2013, 4, 1764. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Dykes, D.; Polesky, H. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef] [PubMed]
- Oskam, C.L.; Haile, J.; McLay, E.; Rigby, P.; Allentoft, M.E.; Olsen, M.E.; Bengtsson, C.; Miller, G.H.; Schwenninger, J.-L.; Jacomb, C. Fossil avian eggshell preserves ancient DNA. Proc. R. Soc. B Biol. Sci. 2010, rspb20092019. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.J.; Llamas, B.; Soubrier, J.; Rawlence, N.J.; Worthy, T.H.; Wood, J.; Lee, M.S.; Cooper, A. Ancient DNA reveals elephant birds and kiwi are sister taxa and clarifies ratite bird evolution. Science 2014, 344, 898–900. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.J.; Wood, J.R.; Scofield, R.P.; Llamas, B.; Cooper, A. Ancient mitochondrial genome reveals unsuspected taxonomic affinity of the extinct Chatham duck (Pachyanas chathamica) and resolves divergence times for New Zealand and sub-Antarctic brown teals. Mol. Phylogenet. Evol. 2014, 70, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.R.; Mitchell, K.J.; Scofield, R.P.; Tennyson, A.; Fidler, A.E.; Wilmshurst, J.M.; Llamas, B.; Cooper, A. An extinct nestorid parrot (Aves, Psittaciformes, Nestoridae) from the Chatham Islands, New Zealand. Zool. J. Linn. Soc. 2014, 172, 185–199. [Google Scholar] [CrossRef]
- Mitchell, K.J.; Wood, J.R.; Llamas, B.; McLenachan, P.A.; Kardailsky, O.; Scofield, R.P.; Worthy, T.H.; Cooper, A. Ancient mitochondrial genomes clarify the evolutionary history of New Zealand’s enigmatic acanthisittid wrens. Mol. Phylogenet. Evol. 2016, 102, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5448. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A. DNA from museum specimens. In Ancient DNA; Springer: New York, NY, USA, 1994; pp. 149–165. [Google Scholar]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Sturrock, S.; Meintjes, P. The Geneious 6.0. 3 Read Mapper; Biomatters Ltd.: Auckland, New Zealand, 2012. [Google Scholar]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M. Analysis of high-throughput ancient DNA sequencing data. In Ancient DNA; Springer: New York, NY, USA, 2012; pp. 197–228. [Google Scholar]
- Jónsson, H.; Ginolhac, A.; Schubert, M.; Johnson, P.L.; Orlando, L. mapDamage2.0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 2013, 29, 1682–1684. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Abbott, C.L.; Double, M.C.; Trueman, J.W.; Robinson, A.; Cockburn, A. An unusual source of apparent mitochondrial heteroplasmy: Duplicate mitochondrial control regions in Thalassarche albatrosses. Mol. Ecol. 2005, 14, 3605–3613. [Google Scholar] [CrossRef] [PubMed]
- García-R, J.C.; Gibb, G.C.; Trewick, S.A. Eocene diversification of crown group rails (Aves: Gruiformes: Rallidae). PLoS ONE 2014, 9, e109635. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, C.; Sipiorski, J.T.; Anderson, F.E. Complete mitochondrial genome sequences and the phylogeny of cranes (Gruiformes: Gruidae). The Auk 2010, 127, 440–452. [Google Scholar] [CrossRef]
- Xia, X. DAMBE6: New tools for microbial genomics, phylogenetics, and molecular evolution. J. Hered. 2017, 108, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Xie, Z. DAMBE: Software package for data analysis in molecular biology and evolution. J. Hered. 2001, 92, 371–373. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Xie, Z.; Salemi, M.; Chen, L.; Wang, Y. An index of substitution saturation and its application. Mol. Phylogenet. Evol. 2003, 26, 1–7. [Google Scholar] [CrossRef]
- Lanfear, R.; Calcott, B.; Ho, S.Y.; Guindon, S. PartitionFinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2016, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Shimodaira, H. An application of multiple comparison techniques to model selection. Ann. Inst. Stat. Math. 1998, 50, 1–13. [Google Scholar] [CrossRef]
- Shimodaira, H.; Hasegawa, M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 1999, 16, 1114. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP*: Phylogenetic Analysis Using Parsimony (and Other Methods), Version 4; Sinauer Associates: Sunderland, MA, USA, 2001.
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Slack, K.E.; Jones, C.M.; Ando, T.; Harrison, G.A.; Fordyce, R.E.; Arnason, U.; Penny, D. Early penguin fossils, plus mitochondrial genomes, calibrate avian evolution. Mol. Biol. Evol. 2006, 23, 1144–1155. [Google Scholar] [CrossRef] [PubMed]
- Weidig, I. New birds from the lower Eocene Green River Formation, North America. Rec. Aust. Mus. 2010, 62, 29–44. [Google Scholar] [CrossRef]
- Feduccia, A.; Voorhies, M.R. Crowned cranes (Gruidae: Balearica) in the Miocene of Nebraska. Nat. Hist. Mus. Los Angel. Cty. Sci. Ser. 1992, 36, 239–248. [Google Scholar]
- Göhlich, U.B. A new crane (Aves: Gruidae) from the Miocene of Germany. J. Vertebr. Paleontol. 2003, 23, 387–393. [Google Scholar] [CrossRef]
- Reddy, S.; Kimball, R.T.; Pandey, A.; Hosner, P.A.; Braun, M.J.; Hackett, S.J.; Han, K.-L.; Harshman, J.; Huddleston, C.J.; Kingston, S.; et al. Why Do Phylogenomic Data Sets Yield Conflicting Trees? Data Type Influences the Avian Tree of Life more than Taxon Sampling. Syst. Biol. 2017, 66, 857–879. [Google Scholar] [CrossRef] [PubMed]
- Stervander, M.; Ryan, P.G.; Melo, M.; Hansson, B. The origin of the world’s smallest flightless bird, the Inaccessible Island Rail Atlantisia rogersi (Aves: Rallidae). Mol. Phylogenet. Evol. 2018, 130, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Slikas, B.; Olson, S.L.; Fleischer, R.C. Rapid, independent evolution of flightlessness in four species of Pacific Island rails (Rallidae): An analysis based on mitochondrial sequence data. J. Avian Biol. 2002, 33, 5–14. [Google Scholar] [CrossRef]
- Gong, J.; Zhao, R.; Huang, Q.; Sun, X.; Huang, L.; Jing, M. Two mitogenomes in Gruiformes (Amaurornis akool/A. phoenicurus) and the phylogenetic placement of Rallidae. Genes Genomics 2017, 39, 987–995. [Google Scholar] [CrossRef]
- Ruan, L.; Wang, Y.; Hu, J.; Ouyang, Y. Polyphyletic origin of the genus Amaurornis inferred from molecular phylogenetic analysis of rails. Biochem. Genet. 2012, 50, 959–966. [Google Scholar] [CrossRef]
- Kirchman, J.J. Speciation of flightless rails on islands: A DNA-based phylogeny of the typical rails of the Pacific—Especiación de Rálidos no Voladores en Islas: Filogenia Basada en ADN de las Formas Típicas del Pacífico. Auk 2012, 129, 56–69. [Google Scholar] [CrossRef]
- Dickinson, E.C.; Bahr, N.; Dowsett, R.; Pearson, D.; Remsen, V.; Roselaar, C.S.; Schodde, D. The Howard and Moore Complete Checklist of Birds of the World, 3rd ed.; A & C Black: London, UK, 2004. [Google Scholar]
- Trewick, S. Flightlessness and phylogeny amongst endemic rails (Aves: Rallidae) of the New Zealand region. Philos. Trans. R. Soc. B Biol. Sci. 1997, 352, 429–446. [Google Scholar] [CrossRef]
- De Pietri, V.L.; Mayr, G. Reappraisal of early Miocene rails (Aves, Rallidae) from central France: Diversity and character evolution. J. Zool. Syst. Evol. Res. 2014, 52, 312–322. [Google Scholar] [CrossRef]
- Ksepka, D.T.; Ware, J.L.; Lamm, K.S. Flying rocks and flying clocks: Disparity in fossil and molecular dates for birds. Philos. Trans. R. Soc. B Biol. Sci. 2014, 281, 2014067. [Google Scholar] [CrossRef] [PubMed]
- Mayr, G. Phylogenetic relationships of the early Tertiary Messel rails (Aves, Messelornithidae). Senckenberg. Lethaea 2004, 84, 317–322. [Google Scholar] [CrossRef]
- Bertelli, S.; Chiappe, L.M.; Mayr, G. A new Messel rail from the Early Eocene Fur Formation of Denmark (Aves, Messelornithidae). J. Syst. Palaeontol. 2011, 9, 551–562. [Google Scholar] [CrossRef]
- Mayr, G. Paleogene Fossil Birds; Springer: Frankfurt, Germany, 2009; ISBN 3-540-89628-7. [Google Scholar]
- Goodman, S.M.; Raherilalao, M.J.; Block, N.L. Patterns of morphological and genetic variation in the Mentocrex kioloides complex (Aves: Gruiformes: Rallidae) from Madagascar, with the description of a new species. Zootaxa 2011, 2776, 49–60. [Google Scholar] [CrossRef]
- Stang, A.T.; McRae, S.B. Why some rails have white tails: The evolution of white undertail plumage and anti-predator signaling. Evol. Ecol. 2009, 23, 943–961. [Google Scholar] [CrossRef]
- Keith, S.; Benson, C.W.; Irwin, M.P.S. The Genus Sarothrura (Aves, Rallidae). Bulletin of the AMNH; v. 143, Article 1; American Museum of Natural History: New York, NY, USA, 1970. [Google Scholar]
- Olson, S.L. Classification of Rallidae. Wilson Bull. 1973, 85, 381–416. [Google Scholar]
- Safford, R.; Hawkins, F. The Birds of Africa: Volume VIII: The Malagasy Region: Madagascar, Seychelles, Comoros, Mascarenes; A & C Black: London, UK, 2013; Volume 8, ISBN 0-7136-6532-7. [Google Scholar]
- Taylor, B. Rails: A Guide to Rails, Crakes, Gallinules and Coots of the World; Christopher Helm Publishers: London, UK, 2010; ISBN 1-4081-3538-8. [Google Scholar]
- Landis, C.; Campbell, H.; Begg, J.; Mildenhall, D.; Paterson, A.M.; Trewick, S. The Waipounamu Erosion Surface: Questioning the antiquity of the New Zealand land surface and terrestrial fauna and flora. Geol. Mag. 2008, 145, 173–197. [Google Scholar] [CrossRef]
- Trewick, S.A. Sympatric flightless rails Gallirallus dieffenbachii and G. modestus on the Chatham Islands, New Zealand; morphometrics and alternative evolutionary scenarios. J. R. Soc. N. Z. 1997, 27, 451–464. [Google Scholar] [CrossRef]
- Trewick, S. Morphology and evolution of two takahe: Flightless rails of New Zealand. J. Zool. 1996, 238, 221–237. [Google Scholar] [CrossRef]
- Askin, R.A.; Spicer, R.A. The Late Cretaceous and Cenozoic history of vegetation and climate at northern and southern high latitudes: A comparison. In Effects of Past Global Change on Life; The National Academies Press: Washington, DC, USA, 1995; pp. 156–173. [Google Scholar]
- Cantrill, D. Early Oligocene Nothofagus from CRP-3, Antarctica: Implications for the vegetation history. Terra Antart. 2001, 8, 401–406. [Google Scholar]
- Mather, E.K.; Tennyson, A.J.; Scofield, R.P.; De Pietri, V.L.; Hand, S.J.; Archer, M.; Handley, W.D.; Worthy, T.H. Flightless rails (Aves: Rallidae) from the early Miocene St Bathans Fauna, Otago, New Zealand. J. Syst. Palaeontol. 2018, 1–27. [Google Scholar] [CrossRef]
- Bunce, M.; Worthy, T.H.; Phillips, M.J.; Holdaway, R.N.; Willerslev, E.; Haile, J.; Shapiro, B.; Scofield, R.P.; Drummond, A.; Kamp, P.J.J.; et al. The evolutionary history of the extinct ratite moa and New Zealand Neogene paleogeography. Proc. Natl. Acad. Sci. USA 2009, 106, 20646–20651. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.B.; Carter, L.; Davey, F.J. The opening of Cook Strait: Interglacial tidal scour and aligning basins at a subduction to transform plate edge. Mar. Geol. 1994, 116, 293–312. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boast, A.P.; Chapman, B.; Herrera, M.B.; Worthy, T.H.; Scofield, R.P.; Tennyson, A.J.D.; Houde, P.; Bunce, M.; Cooper, A.; Mitchell, K.J. Mitochondrial Genomes from New Zealand’s Extinct Adzebills (Aves: Aptornithidae: Aptornis) Support a Sister-Taxon Relationship with the Afro-Madagascan Sarothruridae. Diversity 2019, 11, 24. https://doi.org/10.3390/d11020024
Boast AP, Chapman B, Herrera MB, Worthy TH, Scofield RP, Tennyson AJD, Houde P, Bunce M, Cooper A, Mitchell KJ. Mitochondrial Genomes from New Zealand’s Extinct Adzebills (Aves: Aptornithidae: Aptornis) Support a Sister-Taxon Relationship with the Afro-Madagascan Sarothruridae. Diversity. 2019; 11(2):24. https://doi.org/10.3390/d11020024
Chicago/Turabian StyleBoast, Alexander P., Brendan Chapman, Michael B. Herrera, Trevor H. Worthy, R. Paul Scofield, Alan J. D. Tennyson, Peter Houde, Michael Bunce, Alan Cooper, and Kieren J. Mitchell. 2019. "Mitochondrial Genomes from New Zealand’s Extinct Adzebills (Aves: Aptornithidae: Aptornis) Support a Sister-Taxon Relationship with the Afro-Madagascan Sarothruridae" Diversity 11, no. 2: 24. https://doi.org/10.3390/d11020024
APA StyleBoast, A. P., Chapman, B., Herrera, M. B., Worthy, T. H., Scofield, R. P., Tennyson, A. J. D., Houde, P., Bunce, M., Cooper, A., & Mitchell, K. J. (2019). Mitochondrial Genomes from New Zealand’s Extinct Adzebills (Aves: Aptornithidae: Aptornis) Support a Sister-Taxon Relationship with the Afro-Madagascan Sarothruridae. Diversity, 11(2), 24. https://doi.org/10.3390/d11020024