Microbial Diversity: The Gap between the Estimated and the Known



Abstract
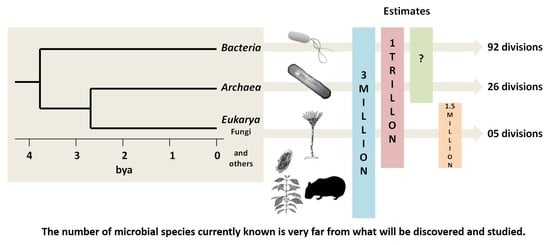
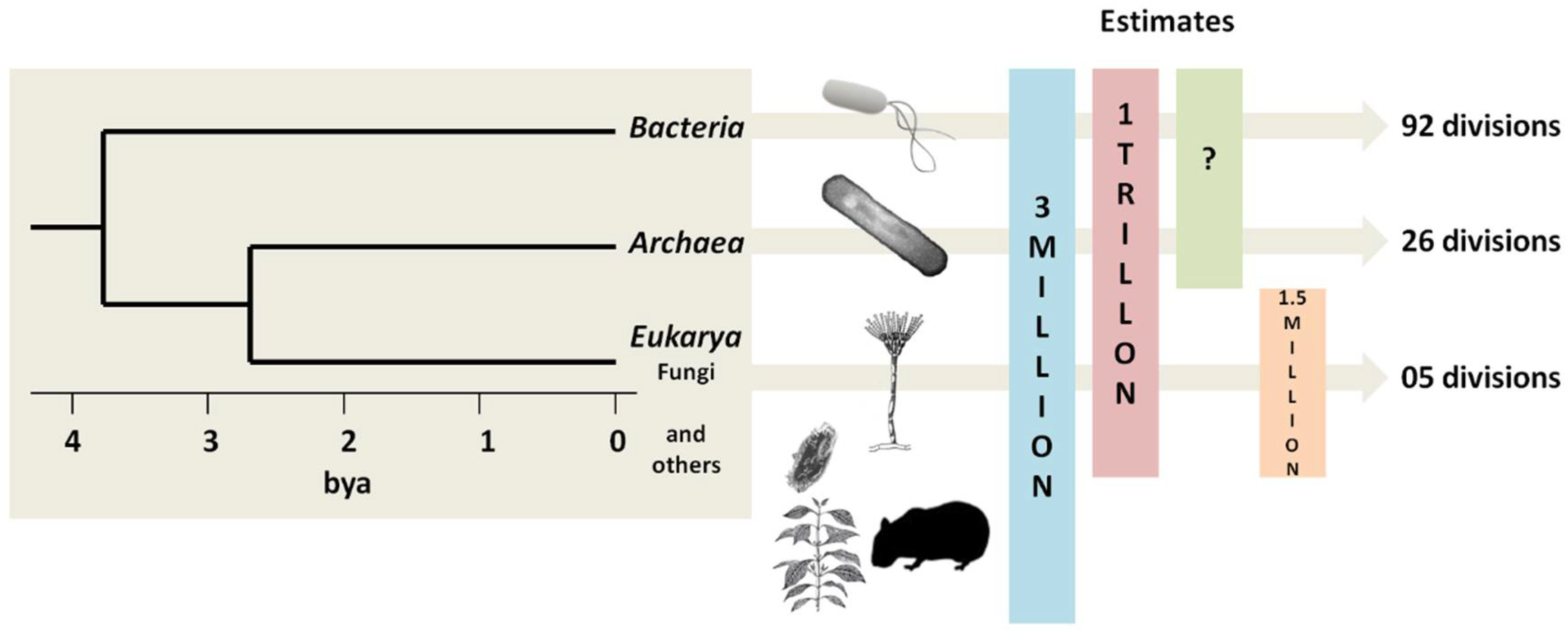
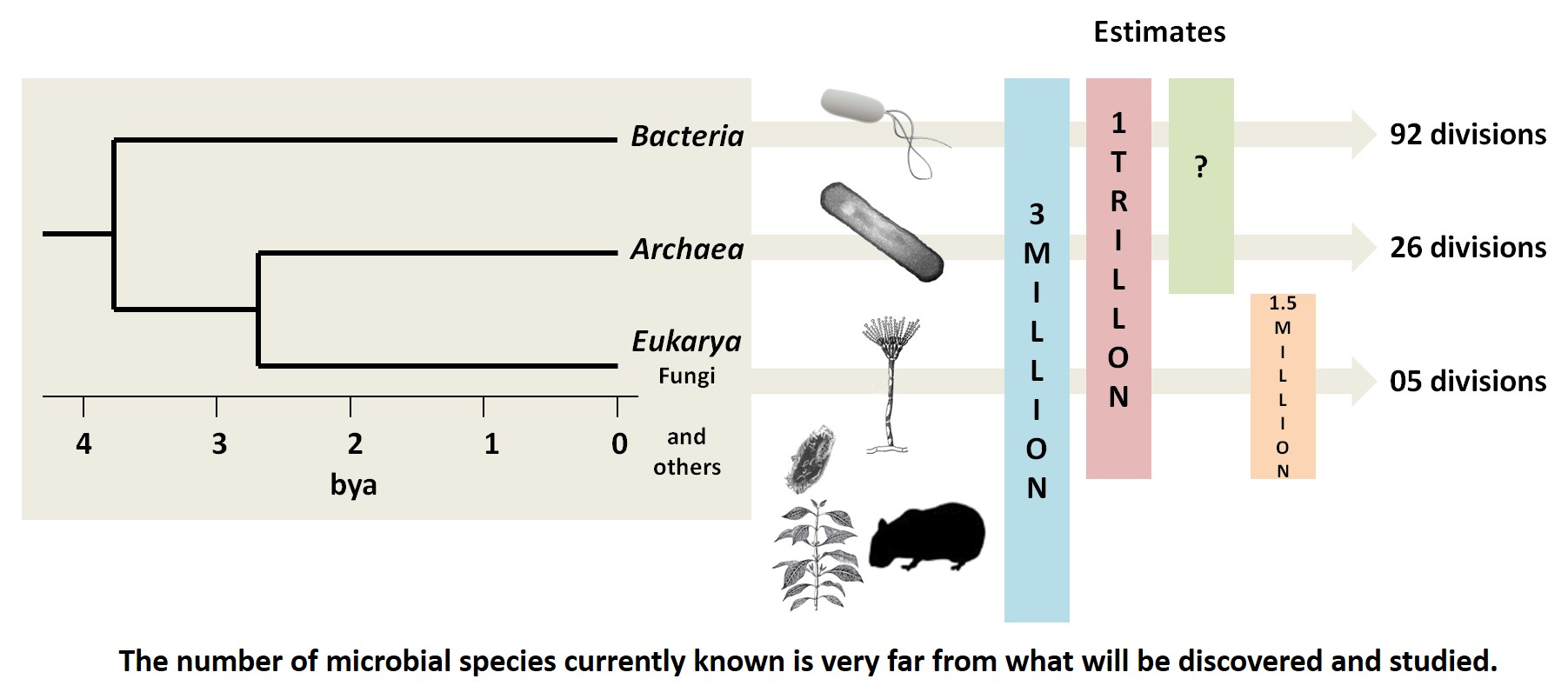{kind=link}
{kind=link}
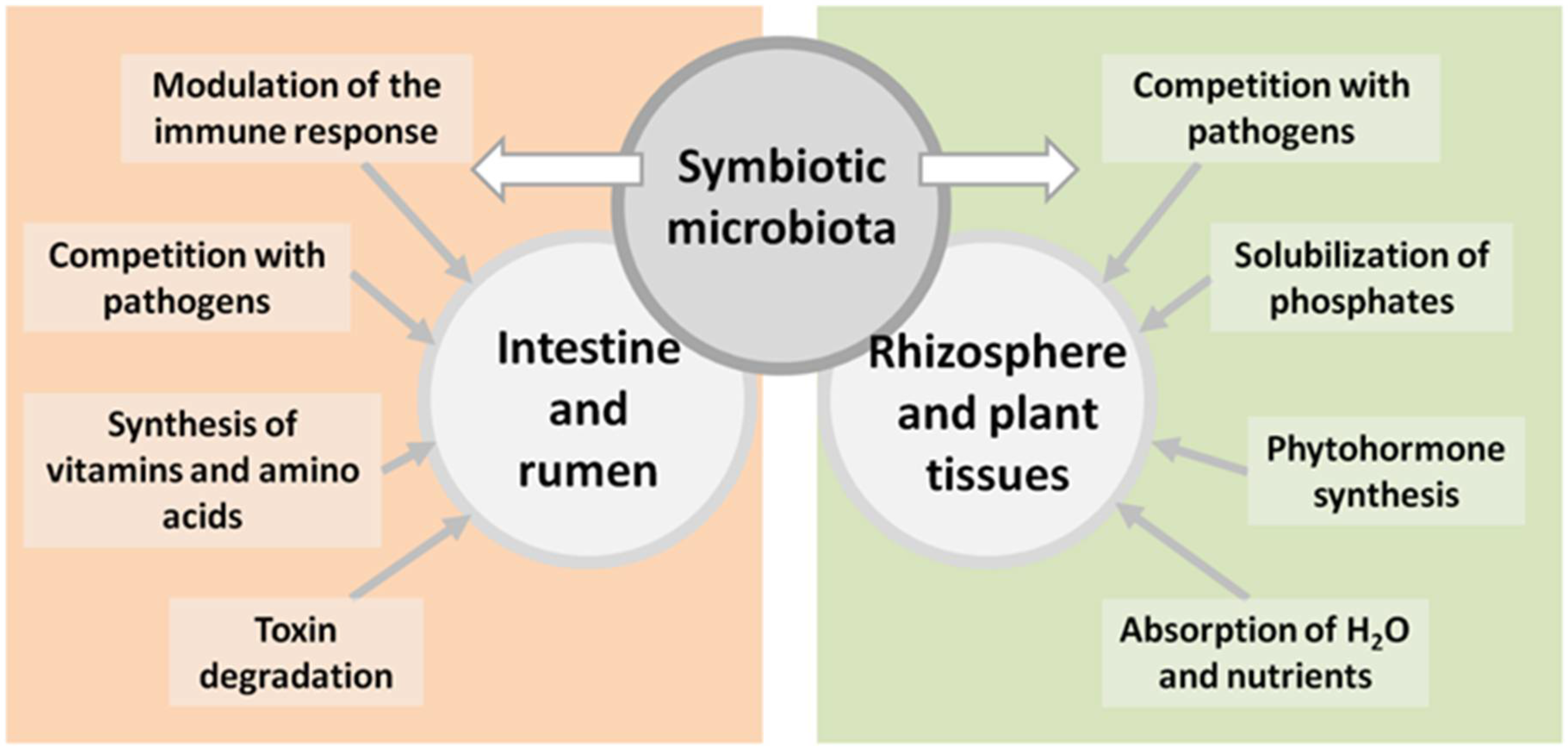{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Biodiversity of the Planet
2. Microbial Diversity: What Do We Have?
3. Why Do We Not Know Microbial Diversity Well?
4. We Do Not See Microorganisms
5. They Cause Diseases
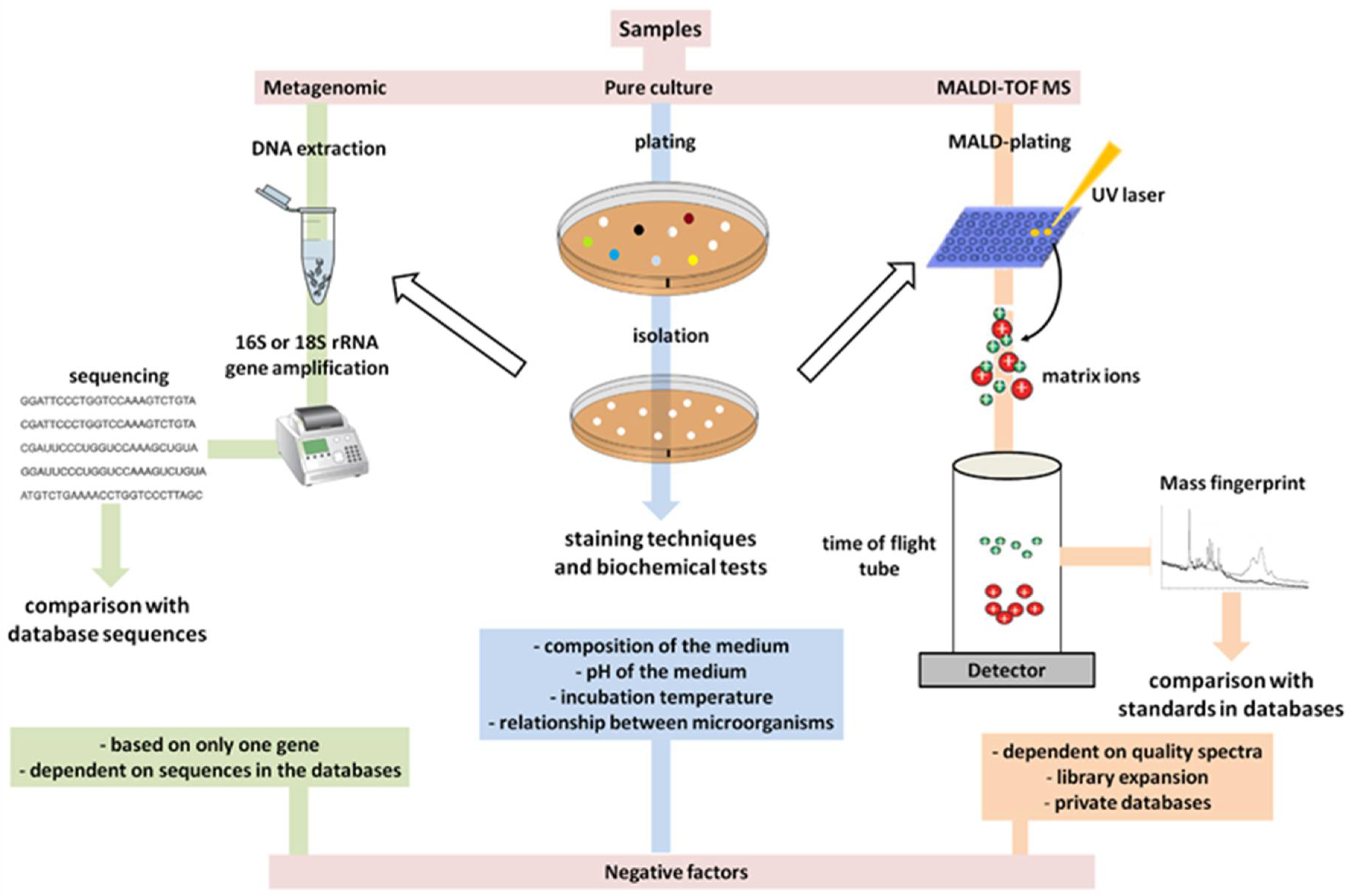
6. The Primary Methods of Detection in Use Have Already Become Outdated
7. Many Are Neither Cosmopolitan or Culturable
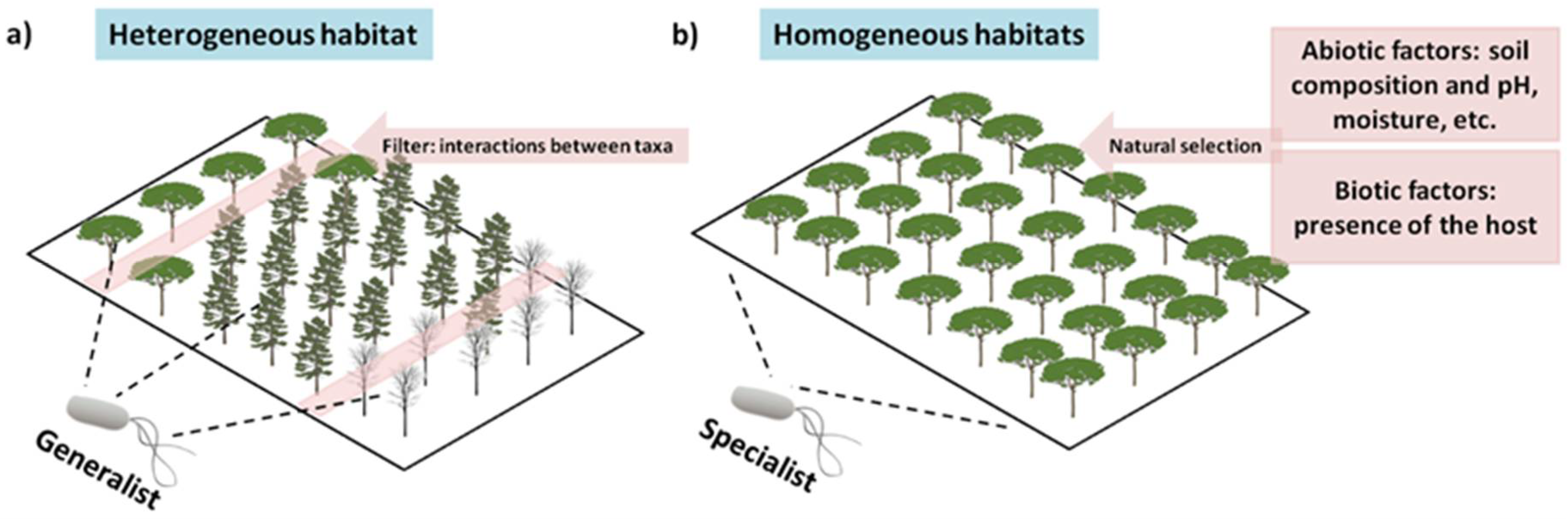
8. They Are Habitat Specialists, and We Have Not yet Evaluated All Habitats
9. Why Should We Know About Microbial Diversity?
10. Additional Observations
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Magurran, A.E. Measuring Biological Diversity; Blackwell: Oxford, UK, 2004; 264p. [Google Scholar]
- Jonsson, P.R.; Hinrichsen, H.H.; Kotta, J.; Kotterba, P.; Middelboe, A.L.; Oesterwind, D.; Bonsdorff, E. Report on the importance of connectivity as a driver of biodiversity (populations, species, communities, habitats). Bonus Bio-C3 2016, 3, 1–27. [Google Scholar] [CrossRef]
- Pomeroy, L.R. The ocean’s food web, a changing paradigm. BioScience 1974, 24, 499–504. [Google Scholar] [CrossRef]
- Azam, F.; Fenchel, T.; Field, J.G.; Gray, J.S.; Meyer-Reil, L.A.; Thingstad, F. The ecological role of water-column microbes in the sea. Mar. Ecol. Prog. Ser. 1983, 10, 257–263. [Google Scholar] [CrossRef]
- Agenda, S. Systematics Agenda 2000: Charting the Biosphere; Technical Report; Systematics Agenda: New York, NY, USA, 1994; pp. 1–34. [Google Scholar]
- Claridge, M.F. Introducing systematic Agenda 2000. Biodivers. Conserv. 1995, 4, 451–452. [Google Scholar] [CrossRef]
- Joppa, L.N.; Roberts, D.L.; Myers, N.; Pimm, S.L. Biodiversity hotspots house most undiscovered plant species. Proc. Natl. Acad. Sci. USA 2011, 108, 13171–13176. [Google Scholar] [CrossRef] [PubMed]
- Mora, C.; Tittensor, D.P.; Adl, S.; Simpson, A.G.B.; Worm, B. How many species are there on earth and in the ocean? PLoS Biol. 2011, 9, e1001127. [Google Scholar] [CrossRef] [PubMed]
- Daly, M.; Herendeen, P.S.; Guralnick, R.P.; Westneat, M.W.; McDade, L. Systematics Agenda 2020: The Mission Evolves. Syst. Biol. 2012, 61, 549–552. [Google Scholar] [CrossRef] [PubMed]
- May, R.M.; Beverton, R.J.H. How many species? Philos. Trans. R. Soc. Lond. B Biol. Sci. 1990, 330, 293–304. [Google Scholar] [CrossRef]
- May, R.M. How many species are there on earth? Science 1988, 241, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Erwin, T.L. Beetles and other insects of tropical forest canopies at Manaus, Brazil, sampled by insecticidal fogging. In Tropical Rainforest: Ecology and Management; Sutton, S.L., Whitmore, T.C., Chadwick, A.C., Eds.; Blackwell Scientific Publications: Oxford, UK, 1983; pp. 59–75. [Google Scholar]
- Raven, P.H. Disappearing species: A global tragedy. Futurist 1985, 19, 8–14. [Google Scholar]
- Stork, N.E. How many species are there? Biodivers. Conserv. 1993, 2, 215–232. [Google Scholar] [CrossRef]
- Reid, W.V.; Miller, K.R. Keeping Options Alive: The Scientific Basis for Conserving Biological Diversity; World Resources Institute: Washington, DC, USA, 1989; 129p. [Google Scholar]
- Reid, W.V. How many species will there be? In Tropical Deforestation and Species Extinction; Whitmore, T.C., Sayer, J.A., Eds.; Chapmam & Hall: London, UK; New York, NY, USA; Tokyo, Japan; Melbourne, Australia; Madras, India, 1992; pp. 55–73. [Google Scholar]
- Simberloff, D. Are we on the verge of a mass extinction in tropical rain forests? In Dynamics of Extinction; Elliott, D.K., Ed.; Wiley: New York, NY, USA, 1986; pp. 165–180. [Google Scholar]
- Dirzo, R.; Raven, P.H. Global state of biodiversity and loss. Annu. Rev. Environ. Resour. 2003, 28, 137–167. [Google Scholar] [CrossRef]
- Gibson, L.; Lee, T.M.; Koh, L.P.; Gardner, T.A.; Barlow, J.; Peres, C.A. Primary forests are irreplaceable for sustaining tropical biodiversity. Nature 2011, 478, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Chazdon, R.L. Beyond deforestation: Restoring forests and ecosystem services on degraded lands. Science 2008, 320, 1458–1460. [Google Scholar] [CrossRef] [PubMed]
- Pereira, H.M. A latitudinal gradient for genetic diversity. Science 2016, 353, 1494–1495. [Google Scholar] [CrossRef] [PubMed]
- Gentry, A.H. Tropical forest biodiversity: Distributional patterns and their conservational significance. Oikos 1992, 63, 19–28. [Google Scholar] [CrossRef]
- Myers, N.; Mittermeier, R.A.; Mittermeier, C.G.; da Fonseca, G.A.; Kent, J. Biodiversity hotspots for conservation priorities. Nature 2000, 403, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Groombridge, B.; Jenkins, M.D. Global Biodiversity: Earth’s Living Resources in the 21st Century; World Conservation Press: Cambridge, UK, 2000; 364p. [Google Scholar]
- Reaka-Kudla, M.L. The global biodiversity of coral reefs: A comparison with rain forests. In Biodiversity II; Reaka-Kudla, M.L., Wilson, D.E., Wilson, E.O., Eds.; Joseph Henry Press: Washington, DC, USA, 1997; pp. 83–108. [Google Scholar]
- Minelli, A. Biological Systematic; Chapman and Hall: London, UK, 1993; 299p. [Google Scholar]
- Hammond, P.M. The current magnitude of biodiversity. In Global Biodiversity Assessment; Heywood, V.H., Watson, R.T., Eds.; Cambridge University Press: Cambridge, UK, 1995; pp. 113–138. [Google Scholar]
- Brusca, R.C.; Brusca, G.J. Invertebrates; Sinauer Associates: Sunderland, MA, USA, 2003; 936p. [Google Scholar]
- Bouchet, P. The magnitude of marine biodiversity. In The Exploration of Marine Biodiversity; Duarte, C.M., Ed.; Fundación BBVA: Bilbao, Spain, 2006; pp. 31–62. [Google Scholar]
- Locey, K.J.; Lennon, J.T. Scaling laws predict global microbial diversity. Proc. Natl. Acad. Sci. USA 2016, 113, 5970–5975. [Google Scholar] [CrossRef] [PubMed]
- Torsvik, V.; Øvreås, L.; Thingstad, T.F. Prokaryotic diversity–magnitude, dynamics, and controlling factors. Science 2002, 296, 1064–1066. [Google Scholar] [CrossRef] [PubMed]
- Gans, J.; Wolinsky, M.; Dunbar, J. Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science 2005, 309, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Roesch, L.F.W.; Fulthorpe, R.R.; Riva, A.; Casella, G.; Hadwin, A.K.; Kent, A.D.; Daroub, S.H.; Camargo, F.A.; Farmerie, W.G.; Triplett, E.W. Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J. 2007, 1, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Curtis, T.P.; Sloan, W.T.; Scannell, J.W. Estimating prokaryotic diversity and its limits. Proc. Natl. Acad. Sci. USA 2002, 99, 10494–10499. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Rainey, F.A. Integrating genomics into the taxonomy and systematics of the Bacteria and Archaea. Int. J. Syst. Evol. Microbiol. 2014, 64, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.O. The Diversity of Life; Harvard University Press: New York, NY, USA, 1994; 440p. [Google Scholar]
- Pace, N.R. A molecular view of microbial diversity and the biosphere. Science 1997, 276, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Ghiorse, W.C. Subterranean life. Science 1997, 275, 789–790. [Google Scholar] [CrossRef]
- Martin, W.F.; Sousa, F.L. Early microbial evolution: The age of anaerobes. Cold Spring Harb. Perspect. Biol. 2015, 8, 2a018127. [Google Scholar] [CrossRef] [PubMed]
- DeLong, E.F.; Pace, N.R. Environmental diversity of bacteria and archaea. Syst. Biol. 2001, 50, 471–478. [Google Scholar] [CrossRef]
- Zahn, L.M. Archaeal diversity and evolution. Science 2017, 357, 560–562. [Google Scholar] [CrossRef]
- Hug, L.A.; Baker, B.J.; Anantharaman, K.; Brown, C.T.; Probst, A.J.; Castelle, C.J.; Butterfield, C.N.; Hernsdorf, A.W.; Amano, Y.; Ise, K.; et al. A new view of the tree of life. Nat. Microbiol. 2016, 16048. [Google Scholar] [CrossRef] [PubMed]
- Dykhuizen, D.E. Santa Rosalia revisited: Why are there so many species of bacteria? Antonie Leeuwenhoek 1998, 73, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Hawksworth, D.L. The fungal dimension of biodiversity: Magnitude, significance, and conservation. Mycol. Res. 1991, 95, 641–655. [Google Scholar] [CrossRef]
- Hawksworth, D.L. The magnitude of fungal diversity: The 1.5 million species estimate revisited. Mycol. Res. 2001, 105, 1422–1432. [Google Scholar] [CrossRef]
- O’Brien, B.L.; Parrent, J.L.; Jackson, J.A.; Moncalvo, J.M.; Vilgalys, R. Fungal community analysis by large-scale sequencing of enviromental samples. Appl. Environ. Microbiol. 2005, 71, 5544–5550. [Google Scholar] [CrossRef] [PubMed]
- Hawksworth, D.L. Global species numbers of fungi: Are tropical studies and molecular approaches contributing to a more robust estimate? Biodivers. Conserv. 2012, 21, 2425–2433. [Google Scholar] [CrossRef]
- Tedersoo, L.; Nara, K. General latitudinal gradient of biodiversity is reversed in ectomycorrhizal fungi. New Phytol. 2010, 185, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Hawksworth, D.L.; Rossman, A.Y. Where are all the undescribed fungi? Phytopathology 1997, 87, 888–891. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, G.S.; Ferrer, A.; Carranza, J. Polypore fungal diversity and host density in a moist tropical forest. Biodivers. Conserv. 2002, 11, 947–957. [Google Scholar] [CrossRef]
- Brundrett, M.C.; Ashwath, N. Glomeromycotan mycorrhizal fungi from tropical Australia III. Measuring diversity in natural and disturbed habitats. Plant Soil 2013, 370, 419–433. [Google Scholar] [CrossRef]
- Schimann, H.; Bach, C.; Lengelle, J.; Louisanna, E.; Barantal, S.; Murat, C.; Buée, M. Diversity and structure of fungal communities in neotropical rainforest soils: The effect of host recurrence. Microb. Ecol. 2017, 73, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Letcher, P.M.; Powell, M.J. Kappamyces a new genus in the Chytridiales (Chytridiomycota). Nova Hedwig. 2005, 80, 115–133. [Google Scholar] [CrossRef]
- Kirk, P.M.; Cannon, P.F.; Minter, D.W.; Stalpers, J.A. Dictionary of the Fungi; CABI: Wallingford, UK, 2008; 784p. [Google Scholar]
- Hibbett, D.S.; Binder, M.; Bischoff, J.F.; Blackwell, M.; Cannon, P.F.; Eriksson, O.E.; Huhndorf, S.; James, T.; Kirk, P.M.; Lücking, R. A higher-level phylogenetic classification of the Fungi. Mycol. Res. 2007, 111, 509–547. [Google Scholar] [CrossRef] [PubMed]
- van der Wal, A.; Geydan, T.D.; Kuyper, T.W.; Boer, W. A thready affair: Linking fungal diversity and community dynamics to terrestrial decomposition processes. FEMS Microbiol. Rev. 2013, 37, 477–494. [Google Scholar] [CrossRef] [PubMed]
- Schüβler, A.; Schwarzott, D.; Walker, C. A new fungal phylum, the Glomeromycota: Phylogeny and evolution. Mycol. Res. 2001, 105, 1413–1421. [Google Scholar] [CrossRef]
- van der Heijden, M.G.A.; Martin, F.M.; Selosse, M.-A.; Sanders, I.R. Mycorrhizal ecology and evolution: The past, the present, and the future. New Phytol. 2015, 205, 1406–1423. [Google Scholar] [CrossRef] [PubMed]
- Öpik, M.; Zobel, M.; Cantero, J.J.; Davison, J.; Facelli, J.M.; Hiiesalu, I.; Jairus, T.; Kalwij, J.M.; Koorem, K.; Leal, M.E. Global sampling of plant roots expands the described molecular diversity of arbuscular mycorrhizal fungi. Mycorrhiza 2013, 23, 411–430. [Google Scholar] [CrossRef] [PubMed]
- Kivlin, S.N.; Hawkes, C.V.; Treseder, K.K. Global diversity and distribution of arbuscular mycorrhizal fungi. Soil Biol. Biochem. 2011, 43, 2294–2303. [Google Scholar] [CrossRef]
- Comandini, O.; Rinaldi, A.C.; Kuyper, T.W. Measuring and estimating ectomycorrhizal fungal diversity: Acontinuous challenge. In Mycorrhiza: Occurrence in Natural and Restored Environments; Pagano, M., Ed.; Nova Science Publishers: New York, NY, USA, 2012; pp. 165–200. [Google Scholar]
- Tedersoo, L.; May, T.W.; Smith, M.E. Ectomycorrhizal lifestyle in fungi: Global diversity, distribution, and evolution of phylogenetic lineages. Mycorrhiza 2010, 20, 217–263. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.F.; Aldrich-Wolfe, L.; Riffel, A.; Barbare, H.; Simpson, N.B.; Trowbridge, J.; Jumpponen, A. Diverse Helotiales associated with the roots of three species of Arctic Ericaceae provide no evidence for host specificity. New Phytol. 2011, 191, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Arnold, A.E.; Maynard, Z.; Gilbert, G.S.; Coley, P.D.; Kursar, T.A. Are tropical fungal endophytes hyperdiverse? Ecol. Lett. 2000, 3, 267–274. [Google Scholar] [CrossRef]
- Arnold, A.E.; Lutzoni, F. Diversity and host range of foliar fungal endophytes: Are tropical leaves biodiversity hotspots? Ecology 2007, 88, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Clay, K.; Shearin, Z.R.C.; Bourke, K.A.; Bickford, W.A.; Kowalski, K.P. Diversity of fungal endophytes in non-native Phragmites australis in the Great Lakes. Biol. Invasions 2016, 18, 2703–2716. [Google Scholar] [CrossRef]
- Shetty, K.G.; Rivadeneira, D.V.; Jayachandran, K. Isolation and molecular characterization of the fungal endophytic microbiome from conventionally and organically grown avocado trees in South Florida. Mycol. Prog. 2016, 15, 977–986. [Google Scholar] [CrossRef]
- Robinson, R.J.; Fraaije, B.A.; Clark, I.M.; Jackson, R.W.; Hirsch, P.R. Endophytic bacterial community composition in wheat (Triticum aestivum) is determined by plant tissue type, developmental stage and soil nutrient availability. Plant Soil 2016, 405, 381–396. [Google Scholar] [CrossRef]
- Blevins, S.M.; Bronze, M.S. Robert Koch and the ‘golden age’ of bacteriology. Int. J. Infect. Dis. 2010, 14, e744–e751. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.L.; Plotkin, S.A. A short history of vaccination. In Vaccines; Plotkin, S.A., Orenstein, W.A., Eds.; Elsevier: Philadelphia, PA, USA, 2004; pp. 1–16. [Google Scholar] [CrossRef]
- Pasteur, L. Summary report of the experiments conducted at Pouilly-le-Fort, near Melun, on the anthrax vaccination, 1881. Yale J. Biol. Med. 2002, 75, 59–62. [Google Scholar] [PubMed]
- Amann, R. Who is out there? Microbial aspects of biodiversity. Syst. Appl. Microbiol. 2000, 23, 1–8. [Google Scholar] [CrossRef]
- White, R.T. The link between the laboratory and learning. Int. J. Sci. Educ. 1996, 18, 761–774. [Google Scholar] [CrossRef]
- Gott, R.; Duggan, S. Practical work: Its role in the understanding of evidence in science. Int. J. Sci. Educ. 1996, 18, 791–806. [Google Scholar] [CrossRef]
- Prokop, P.; Prokop, M.; Tunnicliffe, S.D. Is biology boring? Student attitudes toward biology. J. Biol. Educ. 2007, 42, 36–39. [Google Scholar] [CrossRef]
- Ackert, L. The ‘Cycle of Life’ in Ecology: Sergei Vinogradskii’s Soil Microbiology, 1885–1940. J. Hist. Biol. 2007, 40, 109–145. [Google Scholar] [CrossRef]
- Chung, K.T.; Ferris, D.H. Martinus Willem Beijerinck (1851–1931): Pioneer of general microbiology. ASM News 1996, 62, 539–543. [Google Scholar]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef] [PubMed]
- van Rensburg, J.J.; Dbeibo, L.; Spinola, S.M. The cutaneous microbiota as a determinant of skin barrier function: Molecular interactions and therapeutic opportunities. In Skin Stress Response Pathways; Wondrak, G., Ed.; Springer: Cham, Switzerland, 2016; pp. 379–401. [Google Scholar] [CrossRef]
- Fredricks, D. The Vaginal Microbiome. In Molecular Microbiology; ASM Press: Washington, DC, USA, 2016; pp. 138–145. [Google Scholar]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.; Knight, R.; Gordon, J.I. The human microbiome project: Exploring the microbial part of ourselves in a changing world. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Zárate-Bladés, C.R.; Horai, R.; Caspi, R.R. Regulation of Autoimmunity by the Microbiome. DNA Cell Biol. 2016, 35, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Zmora, N.; Levy, M.; Elinav, E. The microbiome and innate immunity. Nature 2016, 535, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Calo-Mata, P.; Ageitos, J.M.; Böhme, K.; Barros-Velázquez, J. Intestinal microbiota: First barrier against gut-affecting pathogens. In New Weapons to Control Bacterial Growth; Villa, T., Vinas, M., Eds.; Springer: Cham, Switzerland, 2016; pp. 281–314. [Google Scholar] [CrossRef]
- Khoroshkin, M.S.; Rodionov, D. Syntrophic metabolism of vitamins and amino acids in gut microbial community as revealed by in silico genomic analyses. FASEB J. 2016, 30, 819.5. [Google Scholar]
- LeBlanc, J.G.; Chain, F.; Martín, R.; Bermúdez-Humarán, L.G.; Courau, S.; Langella, P. Beneficial effects on host energy metabolism of short-chain fatty acids and vitamins produced by commensal and probiotic bacteria. Microb. Cell Fact. 2017, 16, 79. [Google Scholar] [CrossRef] [PubMed]
- Kamaroff, A.L. The microbiome and risk for obesity and diabetes. JAMA 2017, 317, 355–356. [Google Scholar] [CrossRef] [PubMed]
- Michereff, S.J.; Domingos, E.G.T.; Andrade, M.M. Ecologia e Manejo de Patógenos Radiculares em Solos Tropicais; UFRPE, Imprensa Universitária: Recife, Brazil, 2005; 388p. [Google Scholar]
- Jordan, D.C. Transfer of Rhizobium japonicum Buchanan 1980 to Bradyrhizobium gen. nov., a genus of slow-growing, root nodule bacteria from leguminous plants. Int. J. Syst. Bacteriol. 1982, 32, 136–139. [Google Scholar] [CrossRef]
- He, Z.; Wang, J.; Hu, J.; Zhang, H.; Cai, C.; Shen, J.; Xu, X.; Zheng, P.; Hu, B. Improved PCR primers to amplify 16S rRNA genes from NC10 bacteria. Appl. Microbiol. Biotechnol. 2016, 100, 5099–5108. [Google Scholar] [CrossRef] [PubMed]
- Baker, G.C.; Smith, J.J.; Cowan, D.A. Review and re-analysis of domain-specific 16S primers. J. Microbiol. Methods 2003, 55, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Nübel, U.; Garcia-Pichel, F.; Muyzer, G. PCR primers to amplify 16S rRNA genes from cyanobacteria. Appl. Environ. Microbiol. 1997, 63, 3327–3332. [Google Scholar] [PubMed]
- Tedersoo, L.; Bahram, M.; Puusepp, R.; Nilsson, R.H.; James, T.Y. Novel soil-inhabiting clades fill gaps in the fungal tree of life. Microbiome 2017, 5, 42. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Luo, Z.-H.; Guo, S.; Pang, K.-L. Fungal community analysis in the deep-sea sediments of the Pacific Ocean assessed by comparison of ITS, 18S and 28S ribosomal DNA regions. Deep Sea Res. Part I Oceanogr. Res. Pap. 2016, 109, 51–60. [Google Scholar] [CrossRef]
- Janda, J.M.; Abbott, S.L. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: Pluses, perils, and pitfalls. Clin. Microbiol. 2007, 45, 2761–2764. [Google Scholar] [CrossRef] [PubMed]
- Subrahmanyam, C.S.; Cassidy, B.; Busch, H.; Rothblum, L.I. Nucleotide sequence of the region between the 18S rRNA sequence and the 28S rRNA sequence of rat ribosomal DNA. Nucleic Acids Res. 1982, 10, 3667–3680. [Google Scholar] [CrossRef] [PubMed]
- Rubtsov, P.M.; Musakhanov, M.M.; Zakharyev, V.M.; Krayev, A.S.; Skryabin, K.G.; Bayev, A.A. The structure of the yeast ribosomal RNA genes. I. The complete nucleotide sequence of the 18S ribosomal RNA gene from Saccharomyces cerevisiae. Nucleic Acids Res. 1980, 8, 5779–5794. [Google Scholar] [CrossRef] [PubMed]
- Leal, P.L.; Stürmer, S.L.; Siqueira, J.O. Occurrence and diversity of arbuscular mycorrhizal fungi in trap cultures from soils under different land use systems in the Amazon, Brazil. Braz. J. Microbiol. 2009, 40, 111–121. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Freitas, R.; Buscardo, E.; Nagy, L.; Maciel, A.B.S.; Carrenho, R.; Luizão, R.C.C. Arbuscular mycorrhizal fungal communities along a pedo-hydrological gradient in a Central Amazonian terra firme forest. Mycorrhiza 2014, 24, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, U.; Choudhary, B.K.; Sharma, B.K. Vascular arbuscular mycorrhizal (VAM) spore diversity and density across the soil of degraded forest and rubber plantation in Tripura, India. Am.-Eurasian J. Agric. Environ. Sci. 2014, 14, 1080–1088. [Google Scholar] [CrossRef]
- Husband, R.; Herre, E.A.; Turner, S.L.; Gallery, R.; Young, J.P.W. Molecular diversity of arbuscular mycorrhizal fungi and patterns of host association over time and space in a tropical forest. Mol. Ecol. 2002, 11, 2669–2678. [Google Scholar] [CrossRef] [PubMed]
- Francisco, J.C.; Cohan, F.M.; Krizanc, D. Accuracy and efficiency of algorithms for the demarcation of bacterial ecotypes from DNA sequence data. Int. J. Bioinform. Res. Appl. 2014, 10, 409–425. [Google Scholar] [CrossRef] [PubMed]
- Brochier-Armanet, C.; Forterre, P.; Gribaldo, S. Phylogeny and evolution of the Archaea: One hundred genomes later. Curr. Opin. Microbiol. 2011, 14, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Hugenholtz, P.; Goebel, B.M.; Pace, N.R. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J. Bacteriol. 1998, 180, 4765–4774. [Google Scholar] [PubMed]
- Bogaert, D.; Keijser, B.; Huse, S.; Rossen, J.; Veenhoven, R.; van Gils, E.; Bruin, J.; Montijn, R.; Bonten, M.; Sanders, E. Variability and diversity of nasopharyngeal microbiota in children: A metagenomic analysis. PLoS ONE 2011, 6, e17035. [Google Scholar] [CrossRef] [PubMed]
- Zhernakova, A.; Kurilshikov, A.; Bonder, M.J.; Tigchelaar, E.F.; Schirmer, M.; Vatanen, T.; Mujagic, Z.; Vila, A.V.; Falony, G.; Vieira-Silva, S.; et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 2016, 352, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Wallace, R.J.; Rooke, J.A.; McKain, N.; Duthie, C.-A.; Hyslop, J.J.; Ross, D.W.; Waterhouse, A.; Watson, M.; Roehe, R. The rumen microbial metagenome associated with high methane production in cattle. BMC Genom. 2015, 16, 839. [Google Scholar] [CrossRef] [PubMed]
- Vavourakis, C.D.; Ghai, R.; Rodriguez-Valera, F.; Sorokin, D.Y.; Tringe, S.G.; Hugenholtz, P.; Muyzer, G. Metagenomic insights into the uncultured diversity and physiology of microbes in four hypersaline Soda Lake Brines. Front. Microbiol. 2016, 7, 211. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Chen, J.; Liu, L.; Wu, H.; Tan, H.; Xie, G.; Xu, Q.; Zou, H.; Yu, W.; Wang, L.; Qin, N. Metagenomic sequencing reveals the relationship between microbiota composition and quality of Chinese Rice Wine. Sci. Rep. 2016, 6, 26621. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Breitbart, M.; Nulton, J.; Salamon, P.; Lozupone, C.; Jones, R.; Robeson, M.; Edwards, R.A.; Felts, B.; Rayhawk, S.; et al. Metagenomic and small-subunit rRNA analyses reveal the genetic diversity of bacteria, archaea, fungi, and viruses in soil. Appl. Environ. Microbiol. 2007, 73, 7059–7066. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, M.; Werner, J.; Chernikova, T.N.; Bargiela, R.; Fernández, L.; La Cono, V.; Waldmann, J.; Teeling, J.; Golyshina, O.V.; Glöckner, F.O.; et al. Unveiling microbial life in the new deep-sea hypersaline Lake Thetis. Part II: A metagenomic study. Environ. Microbiol. 2012, 14, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Debroas, D.; Humbert, J.-F.; Enault, F.; Bronner, G.; Faubladier, M.; Cornillor, E. Metagenomic approach studying the taxonomic and functional diversity of the bacterial community in a mesotrophic lake (Lac du Bourget–France). Environ. Microbiol. 2009, 11, 2412–2424. [Google Scholar] [CrossRef] [PubMed]
- Nacke, H.; Goldmann, K.; Schöning, I.; Pfeiffer, B.; Kaiser, K.; Castillo-Villamizar, G.A. Fine spatial scale variation of soil microbial communities under European beech and Norway spruce. Front. Microbiol. 2016, 7, 2067. [Google Scholar] [CrossRef] [PubMed]
- Mašínová, T.; Bahnmann, B.D.; Větrovský, T.; Tomšovský, M.; Merunková, K.; Baldrian, P. Drivers of yeast community composition in the litter and soil of a temperate forest. FEMS Microbiol. Ecol. 2017, 93, fiw223. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-T.; Whitman, W.B.; Coleman, D.C.; Jien, S.-H.; Chiu, C.-Y. Cedar and bamboo plantations alter structure and diversity of the soil bacterial community from a hardwood forest in subtropical mountain. Appl. Soil Ecol. 2017, 112, 28–33. [Google Scholar] [CrossRef]
- Sheldrake, M.; Rosenstock, N.P.; Revillini, D.; Olsson, P.A.; Mangan, S.; Sayer, E.J.; Wallander, H.; Turner, B.L.; Tanner, E.V.J. Arbuscular mycorrhizal fungal community composition is altered by long-term litter removal but not litter addition in a lowland tropical forest. New Phytol. 2017, 214, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Sanders, I.R.; Rodriguez, A. Aligning molecular studies of mycorrhizal fungal diversity with ecologically important levels of diversity in ecosystems. ISME J. 2016, 10, 2780–2786. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, B.P.; Staley, J.T. Microbial endemism and biogeography. In Microbial Diversity and Bioprospecting; Bull, A.T., Ed.; ASM Press: Washington, DC, USA, 2004; pp. 225–231. [Google Scholar]
- Green, J.; Bohannan, B.J.M. Spatial scaling of microbial biodiversity. Microb. Ecol. 2006, 21, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, J. Novas metodologias de identificação de micro-organismos: MALDI-TOF. Einstein 2012, 10, 118–119. [Google Scholar] [CrossRef] [PubMed]
- Benagli, C.; Rossi, V.; Dolina, M.; Tonolla, M.; Petrini, O. Matrix-assisted laser desorption ionization-time of flight mass spectrometry for the identification of clinically relevant bacteria. PLoS ONE 2011, 6, e16424. [Google Scholar] [CrossRef] [PubMed]
- Neville, S.A.; LeCordier, A.; Ziochos, H.; Chater, M.J.; Gosbell, I.B.; Maley, M.W.; van Hal, S.J. The utility of matrix assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) following introduction for routine laboratory bacterial identification. J. Clin. Microbiol. 2011, 49, 2980–2984. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, S.-Y.; Tseng, C.-L.; Lee, Y.-S.; Kuo, A.-J.; Sun, C.-F.; Lin, Y.-H.; Chen, J.-K. Highly efficient classification and identification of human pathogenic bacteria by MALDI-TOF MS. Mol. Cell Proteom. 2008, 7, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Opota, O.; Prod’hom, G.; Greub, G. Applications of MALDI-TOF mass spectrometry in clinical diagnostic microbiology. In Maldi-Tof and Tandem MS for Clinical Microbiology; Shah, H.N., Gharbia, S.E., Eds.; Wiley: New York, NY, USA, 2017; pp. 55–92. [Google Scholar] [CrossRef]
- Siller-Ruiz, M.; Hernández-Egido, S.; Sánchez-Juanes, F.; González-Buitrago, J.M.; Muñoz-Bellido, J.L. Fast methods of fungal and bacterial identification. MALDI-TOF mass spectrometry, chromogenic media. Enferm. Infecc. Microbiol. Clin. 2017, 35, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Kostrzewa, M.; Nagy, E. How MALDI-TOF mass spectrometry can aid diagnosis of hard-to-identify pathogenic bacteria. Expert Rev. Mol. Diagn. 2016, 16, 509–511. [Google Scholar] [CrossRef] [PubMed]
- Lasch, P.; Grunow, R.; Antonation, K.; Weller, S.A.; Jacob, D. Inactivation techniques for MALDI-TOF MS analysis of highly pathogenic bacteria—A critical review. Trends Anal. Chem. 2016, 85, 112–119. [Google Scholar] [CrossRef]
- Lasch, P.; Jacob, D.; Grunow, R.; Schwecke, T.; Doellinger, J. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry (MS) for the identification of highly pathogenic bacteria. Trends Anal. Chem. 2016, 85, 103–111. [Google Scholar] [CrossRef]
- Bader, O. Fungal species identification by MALDI-TOF mass spectrometry. Methods Mol. Biol. 2017, 1508, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Popović, N.T.; Kazazić, S.P.; Strunjak-Perović, I.; Čož-Rakovac, R. Differentiation of environmental aquatic bacterial isolates by MALDI-TOF MS. Environ. Res. 2017, 152, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Molina, E.; Juanes, F.S.; Carro, L.; Flores-Félix, J.D.; Martínez-Hidalgo, P.; Castillo, E.C.; Buitrago, J.M.G.; Velázquez, E. Identification of rhizobial strains nodulating Pisum sativum in northern spain soils by MALDI-TOF MS (Matrix-assisted laser desorption ionization time-of-flight mass spectrometry) analysis. In Biological Nitrogen Fixation and Beneficial Plant-Microbe Interaction; González-Andrés, F., James, E., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 37–44. [Google Scholar] [CrossRef]
- Urquiza, C.C.; Hernández, I.A.; Medina, J.A.C.; López, M.A.R.; Cruz, J.P.; Zarazúa, R.L.R. Identification by MALDI-TOF mass spectrometry of mercury-resistant bacteria associated with the rhizosphere of an apple orchard. Geomicrobiol. J. 2016, 34, 176–182. [Google Scholar] [CrossRef]
- Avanzi, I.R.; Gracioso, L.H.; Baltazar, M.P.G.; Karolski, B.; Perpetuo, E.A.; Nascimento, C.A.O. Rapid bacteria identification from environmental mining samples using MALDI-TOF MS analysis. Environ. Sci. Pollut. Res. Int. 2017, 24, 3717–3726. [Google Scholar] [CrossRef] [PubMed]
- Finlay, B.J.; Clarke, K.J. Ubiquitous dispersal of microbial species. Nature 1999, 400, 828. [Google Scholar] [CrossRef]
- Shurin, J.B.; Cottenie, K.; Hillebrand, H. Autocorrelação espacial e limitação da dispersão em organismos de água doce. Oecologia 2009, 159, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Fenchel, T.; Finlay, B.J. The ubiquity of small species: Patterns of local and global diversity. BioScience 2004, 54, 777–784. [Google Scholar] [CrossRef]
- Papke, R.T.; Ramsing, N.B.; Bateson, M.M.; Ward, D.M. Geographical isolation in hot spring cyanobacteria. Environ. Microbiol. 2003, 5, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Papke, R.T.; Ward, D.M. The importance of physical isolation to microbial diversification. FEMS Microbiol. Ecol. 2004, 48, 293–303. [Google Scholar] [CrossRef] [PubMed]
- de Wit, R.; Bouvier, T. ‘Everything is everywhere, but, the environment selects’; what did Baas Becking and Beijerinck really say? Environ. Microbiol. 2006, 8, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Martiny, J.B.; Bohannan, B.J.; Brown, J.H.; Colwell, R.K.; Fuhrman, J.A.; Green, J.L.; Horner-Devine, M.C.; Kane, M.; Krumins, J.A.; Kuske, C.R.; et al. Microbial biogeography: Putting microorganisms on the map. Nat. Rev. Microbiol. 2006, 4, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Telford, R.J.; Vandvik, V.; Birks, H.J.B. Dispersal limitations matter for microbial morphospecies. Science 2006, 312, 1015. [Google Scholar] [CrossRef] [PubMed]
- Hanson, C.; Fuhrman, J.; Horner-Devine, M.C.; Martiny, J.B.H. Beyond biogeographic patterns: Processes shaping the microbial landscape. Nat. Rev. Microbiol. 2012, 10, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Tamames, J.; Abellan, J.J.; Pignatelli, M.; Camacho, A.; Moya, A. Environmental distribution of prokaryotic taxa. BMC Microbiol. 2010, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.W.; Koll, U.; Jezberová, J.; Camacho, A. Global phylogeography of pelagic Polynucleobacter bacteria: Restricted geographic distribution of subgroups, isolation by distance and influence of climate. Environ. Microbiol. 2015, 17, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Liu, Y.; Hu, A.; Liu, X.; Chen, F.; Yao, T. Genetic diversity of picocyanobacteria in Tibetan lakes: Assessing the endemic and universal distributions. Appl. Environ. Microbiol. 2014, 80, 7640–7650. [Google Scholar] [CrossRef] [PubMed]
- Oda, Y.; Star, B.; Huisman, L.A. Biogeography of the purple nonsulfur bacterium Rhodopseudomonas palustris. Appl. Environ. Microbiol. 2003, 69, 5186–5191. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, R.J.; Grogan, D.W.; Taylor, J.W. Geographic barriers isolate endemic populations of hyperthermophilic archaea. Science 2003, 301, 976–978. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.-C.; Tiedje, J.M. Biogeography and degree of endemicity of fluorescent Pseudomonas strains in soil. Appl. Environ. Microbiol. 2000, 66, 5448–5456. [Google Scholar] [CrossRef] [PubMed]
- van Tol, H.M.; Amin, S.A.; Armbrust, E.V. Ubiquitous marine bacterium inhibits diatom cell division. ISME J. 2017, 11, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Schneiker, S.; Santos, V.A.P.M.; Bartels, D.; Bekel, T.; Brecht, M.; Buhrmester, J.; Chernikova, T.N.; Denaro, R.; Ferrer, M.; Gertler, C.; et al. Genome sequence of the ubiquitous hydrocarbon-degrading marine bacterium Alcanivorax borkumensis. Nat. Biotechnol. 2006, 24, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Rappé, M.S.; Connon, S.A.; Vergin, K.L.; Giovannoni, S.J. Cultivation of the ubiquitous SAR11 marine bacterioplankton clade. Nature 2002, 418, 630–633. [Google Scholar] [CrossRef] [PubMed]
- Noguez, A.M.; Arita, H.T.; Escalente, A.E.; Forney, L.J.; García-Oliva, F.; Souza, V. Microbial macroecology: Highly structured prokaryotic soil assemblages in a tropical deciduous forest. Glob. Ecol. Biogeogr. 2005, 14, 241–248. [Google Scholar] [CrossRef]
- Garcia, S.L. Mixed cultures as model communities: Hunting for ubiquitous microorganisms, their partners, and interactions. Aquat. Microb. Ecol. 2016, 77, 79–85. [Google Scholar] [CrossRef]
- Mendes, R.; Garbeva, P.; Raaijmakers, J.M. The rhizosphere microbiome: Significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol. Rev. 2013, 37, 634–663. [Google Scholar] [CrossRef] [PubMed]
- Ward, B.B. How many species of prokaryotes are there? Proc. Natl. Acad. Sci. USA 2002, 99, 10234–10236. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 2013, 12, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schäberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Logares, R.; Lindström, E.S.; Langenheder, S.; Logue, J.B.; Paterson, H.; Laybourn-Parry, J.; Rengefors, K.; Tranvik, L.; Bertilsson, S. Biogeography of bacterial communities exposed to progressive long-term environmental change. ISME J. 2013, 7, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Juutilainen, K.; Mönkkönen, M.; Kotiranta, H.; Halme, P. Resource use of wood-inhabiting fungi in different boreal forest types. Fungal Ecol. 2017, 27, 96–106. [Google Scholar] [CrossRef]
- Kassen, R. The experimental evolution of specialists, generalists, and the maintenance of diversity. J. Evol. Biol. 2002, 15, 173–190. [Google Scholar] [CrossRef]
- Comte, J.; Lindström, E.S.; Eiler, A.; Langenheder, S. Can marine bacteria be recruited from freshwater sources and the air? ISME J. 2014, 8, 2423–2430. [Google Scholar] [CrossRef] [PubMed]
- Wehner, J.; Mittelbach, M.; Rillig, M.C.; Verbruggen, E. Specialist nectar-yeasts decline with urbanization in Berlin. Sci. Rep. 2017, 7, 45315. [Google Scholar] [CrossRef] [PubMed]
- Székely, A.J.; Langenheder, S. The importance of species sorting differs between habitat generalists and specialists in bacterial communities. FEMS Microbiol. Ecol. 2014, 87, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Székely, A.J.; Berga, M.; Langenheder, S. Mechanisms determining the fate of dispersed bacterial communities in new environments. ISME J. 2013, 7, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.A.; Nara, K.; Hogetsu, T. Host effects on ectomycorrhizal fungal communities: Insight from eight host species in mixed conifer-broadleaf forests. New Phytol. 2007, 174, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Kernaghan, G.; Patriquin, G. Host associations between fungal root endophytes and boreal trees. Microb. Ecol. 2011, 62, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Tedersoo, L.; Mett, M.; Ishida, T.A.; Bahram, M. Phylogenetic relationships among host plants explain differences in fungal species richness and community composition in ectomycorrhizal symbiosis. New Phytol. 2013, 199, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Bálint, M.; Bartha, L.; O’Hara, R.; Olson, M.S.; Otte, J.; Pfenninger, M.; Pfenninger, M.; Robertson, A.L.; Tiffin, P.; Schmitt, I. Relocation, high-latitude warming and host genetic identity shape the foliar fungal microbiome of poplars. Mol. Ecol. 2015, 24, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liang, M.; Etienne, R.S.; Wang, Y.; Staehelin, C.; Yu, S. Experimental evidence for a phylogenetic Janzen-Connell effect in a subtropical forest. Ecol. Lett. 2011, 15, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Daniell, T.J.; Husband, R.; Fitter, A.H.; Young, J.P.W. Molecular diversity of arbuscular mycorrhizal fungi colonising arable crops. FEMS Microbiol. Ecol. 2001, 36, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Vandenkoornhuyse, P.; Husband, R.; Daniell, T.J.; Watson, I.J.; Duck, J.M.; Fitter, A.H.; Young, J.P. Arbuscular mycorrhizal community composition associated with two plant species in a grassland ecosystem. Mol. Ecol. 2002, 11, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Elster, J.; Margesin, R.; Wagner, D.; Häggblom, M. Editorial: Polar and Alpine Microbiology-Earth’s cryobiosphere. FEMS Microbiol. Ecol. 2017, 93, fiw221. [Google Scholar] [CrossRef] [PubMed]
- Aliyu, H.; De Maayer, P.; Cowan, D. The genome of the Antarctic polyextremophile Nesterenkonia sp. AN1 reveals adaptive strategies for survival under multiple stress conditions. FEMS Microbiol. Ecol. 2016, 92, fiw032. [Google Scholar] [CrossRef] [PubMed]
- Goordial, J.; Raymond-Bouchard, I.; Zolotarov, Y.; de Bethencourt, L.; Ronholm, J.; Shapiro, N.; Woyke, T.; Stromvik, M.; Greer, C.W.; Bakermans, C.; et al. Cold adaptive traits revealed by comparative genomic analysis of the eurypsychrophile Rhodococcus sp. JG3 isolated from high elevation McMurdo Dry Valley permafrost, Antarctica. FEMS Microbiol. Ecol. 2016, 92, fiv154. [Google Scholar] [CrossRef] [PubMed]
- Petrovskaya, L.E.; Novotoskaya-Vlasova, K.A.; Spirina, E.V.; Durdenko, E.V.; Lomakina, G.Y.; Zavialova, M.G.; Nikolaev, E.N.; Rivkina, E.M. Expression and characterization of a new esterase with GCSAG motif from a permafrost metagenomic library. FEMS Microbiol. Ecol. 2016, 92, fiw046. [Google Scholar] [CrossRef] [PubMed]
- Ciok, A.; Dziewit, L.; Grzesiak, J.; Budzik, K.; Gorniak, D.; Zdanowski, M.K.; Bartosik, D. Identification of miniature plasmids in psychrophilic Arctic bacteria of the genus Variovorax. FEMS Microbiol. Ecol. 2016, 92, fiw043. [Google Scholar] [CrossRef] [PubMed]
- Baulina, O.; Gorelova, O.; Solovchenko, A.; Chivkunova, O.; Semenova, L.; Selyakh, I.; Scherbakov, P.; Burakova, O.; Lobakova, E. Diversity of the nitrogen starvation responses in subarctic Desmodesmus sp. (Chlorophyceae) strains isolated from symbioses with invertebrates. FEMS Microbiol. Ecol. 2016, 92, fiw031. [Google Scholar] [CrossRef] [PubMed]
- Herburger, K.; Remias, D.; Holzinger, A. The green alga Zygogonium ericetorum (Zygnematophyceae, Charophyta) shows high iron and aluminium tolerance: Protection mechanisms and photosynthetic performance. FEMS Microbiol. Ecol. 2016, 92, fiw103. [Google Scholar] [CrossRef] [PubMed]
- Remias, D.; Pichrtová, M.; Pangratz, M.; Lütz, C.; Holzinger, A. Ecophysiology, secondary pigments and ultrastructure of Chlainomonas sp. (Chlorophyta) from the European Alps compared with Chlamydomonas nivalis forming red snow. FEMS Microbiol. Ecol. 2016, 92, fiw030. [Google Scholar] [CrossRef] [PubMed]
- Colangelo-Lillis, J.; Eicken, H.; Carpenter, S.D.; Deming, J.W. Evidence for marine origin and microbial-viral habitability of sub-zero hypersaline aqueous inclusions within permafrost near Barrow, Alaska. FEMS Microbiol. Ecol. 2016, 92, fiw053. [Google Scholar] [CrossRef] [PubMed]
- de Cárcer, D.A.; López-Bueno, A.; Alonso-Lobo, J.M.; Quesada, A.; Alcamí, A. Metagenomic analysis of lacustrine viral diversity along a latitudinal transect of the Antarctic Peninsula. FEMS Microbiol. Ecol. 2016, 92, fiw074. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.; Ahmed, I.; Salam, N.; Kim, B.-Y.; Singh, D.; Zhi, X.-Y.; Xiao, M.; Li, W.J. Diversity and distribution of thermophilic bacteria in hot springs of Pakistan. Microb. Ecol. 2017, 74, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Hou, W.; Dong, H.; Jiang, H.; Huang, L.; Wu, G.; Zhang, C.; Song, Z.; Zhang, Y.; Ren, H.; et al. Control of temperature on microbial community structure in hot springs of the Tibetan plateau. PLoS ONE 2013, 8, e62901. [Google Scholar] [CrossRef] [PubMed]
- Kubo, K.; Knittel, K.; Amann, R.; Fukui, M.; Matsuura, K. Sulfur-metabolizing bacterial populations in microbial mats of the Nakabusa hot spring, Japan. Syst. Appl. Microbiol. 2011, 34, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Bohorquez, L.C.; Delgado-Serrano, L.; Lopez, G.; Osorio-Forero, C.; Klepac-Ceraj, V.; Kolter, R.; Junca, H.; Baena, S.; Zambrano, M.M. In-depth characterization via complementing culture-independent approaches of microbial community in an acidic hot spring of the Colombian Andes. Microb. Ecol. 2012, 63, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Kanoksilapatham, W.; Pasomsup, P.; Keawram, P.; Cuecas, A.; Portillo, M.C.; Gonzalez, J.M. Fervidobacterium thailandense sp. nov., an extremely thermophilic bacterium isolated from a hot spring. Int. J. Syst. Evol. Microbiol. 2016, 66, 5023–5027. [Google Scholar] [CrossRef] [PubMed]
- Ming, H.; Duan, Y.-Y.; Yin, Y.-R.; Meng, X.-L.; Li, S.; Zhou, E.-M.; Huang, J.R.; Nie, G.X.; Li, W.J. Crenalkalicoccus roseus gen. nov., sp. nov., a thermophilic bacterium isolated from alkaline hot springs. Int. J. Syst. Evol. Microbiol. 2016, 66, 2319–2326. [Google Scholar] [CrossRef] [PubMed]
- Priya, I.; Dhar, M.K.; Bajaj, B.K.; Koul, S.; Vakhlu, J. Cellulolytic activity of thermophilic bacilli isolated from Tattapani hot spring sediment in north west Himalayas. Indian J. Microbiol. 2016, 56, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Tank, M.; Thiel, V.; Ward, D.M.; Bryant, D.A. A panoply of phototrophs: An overview of the thermophilic chlorophototrophs of the microbial mats of alkaline siliceous hot springs in Yellowstone national park, WY, USA. In Modern Topics in the Phototrophic Prokaryotes; Hallenbeck, P.C., Ed.; Springer: Cham, Switzerland, 2017; pp. 87–137. [Google Scholar] [CrossRef]
- Tazi, L.; Breakwell, D.P.; Harker, A.R.; Crandall, K.A. Life in extreme environments: Microbial diversity in Great Salt Lake, Utah. Extremophiles 2014, 18, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, M.B.; Karray, F.; Mhiri, N.; Mei, N.; Quéméneur, M.; Cayol, J.-L.; Erauso, G.; Tholozan, J.L.; Alazard, D.; Sayadi, S. Prokaryotic diversity in a Tunisian hypersaline lake, Chott El Jerid. Extremophiles 2016, 20, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Khalilova, E.A.; Kotenko, S.T.; Islammagomedova, E.A.; Gasanov, R.Z.; Abakarova, A.A.; Aliverdieva, D.A. Extremophilic microbial communities of saline soils and their diversity in the regions of the Caspian Depression. Arid Ecosyst. 2017, 7, 116–120. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Liang, J.; Song, Q.; Zhang, X.-H. Degradation properties of various macromolecules of culturable psychrophilic bacteria from the deep-sea water of the South Pacific Gyre. Extremophiles 2016, 20, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Pathom-aree, W.; Stach, J.E.M.; Ward, A.C.; Horikoshi, K.; Bull, A.T.; Goodfellow, M. Diversity of actinomycetes isolated from Challenger Deep sediment (10,898 m) from the Mariana Trench. Extremophiles 2006, 10, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Jebbar, M. Deep sea, the last great unexplored Earth frontier harboring the largest unknown and untapped remote microbial diversity on our planet. Res. Microbiol. 2015, 166, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Lauro, F.M.; Bartlett, D.H. Prokaryotic lifestyles in deep sea habitats. Extremophiles 2008, 12, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Musilova, M.; Wright, G.; Ward, J.M.; Dartnell, L.R. Isolation of radiation-resistant bacteria from mars analog Antarctic dry valleys by preselection, and the correlation between radiation and desiccation resistance. Astrobiology 2015, 15, 1076–1090. [Google Scholar] [CrossRef] [PubMed]
- Albarracín, V.H.; Simon, J.; Pathak, G.P.; Valle, L.; Douki, T.; Cadet, J.; Borsarelli, C.D.; Farias, M.E.; Gärtner, W. First characterisation of a CPD-class I photolyase from a UV-resistant extremophile isolated from High-Altitude Andean Lakes. Photochem. Photobiol. Sci. 2014, 13, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Morozova, D.; Moeller, R.; Rettberg, P.; Wagner, D. Enhanced radiation resistance of Methanosarcina soligelidi SMA-21, a new methanogenic archaeon isolated from a siberian permafrost-affected soil in direct comparison to Methanosarcina barkeri. Astrobiology 2015, 15, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Chan, O.W.; Lacap-Bugler, D.C.; Pointing, S.B. Radiation-tolerant bacteria isolated from high altitude soil in Tibet. Indian J. Microbiol. 2016, 56, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Krisko, A.; Radman, M. Biology of Extreme Radiation Resistance: The Way of Deinococcus radiodurans. Cold Spring Harb. Perspect. Biol. 2013, 5, a012765. [Google Scholar] [CrossRef] [PubMed]
- Vitorino, L.C.; Bessa, L.A. Technological microbiology: Development and applications. Front. Microbiol. 2017, 8, 827. [Google Scholar] [CrossRef] [PubMed]
- Vlasova, A.N.; Kandasamy, S.; Chattha, K.S.; Rajashekara, G.; Saif, L.J. Comparison of probiotic lactobacilli and bifidobacteria effects, immune responses and rotavirus vaccines and infection in different host species. Vet. Immunol. Immunopathol. 2016, 172, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Warehan, K.; Wang, D.; Bradley, C.; Hutchings, H.; Harris, W.; Dhar, A.; Brown, H.; Foden, A.; Gravenor, M.B.; et al. Lactobacilli and bifidobacteria in the prevention of antibiotic-associated diarrhoea and Clostridium difficile diarrhoea in older inpatients (PLACIDE): A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2013, 1249–1257. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, J.; Guo, Z.; Kwok, L.; Ma, C.; Zhang, W.; Lv, Q.; Huang, W.; Zhang, H. Effect of oral consumption of probiotic Lactobacillus planatarum P-8 on fecal microbiota, SIgA, SCFAs, and TBAs of adults of different ages. Nutrition 2014, 30, 776–783.e1. [Google Scholar] [CrossRef] [PubMed]
- Al-Otaibi, H.S.; Gashgari, R.M.; Mohammed, A.E.; Almojel, A.S.; Elobeid, M.M.; Abrahaim, J.S.A. Investigation of the growth ability of probiotic (Lactobacillus and Bifidobacterium) in infant’s milk under different environmental conditions. Biomed. Pharmacol. J. 2016, 9, 451–462. [Google Scholar] [CrossRef]
- Lodemann, U.; Strahlendorf, J.; Schierack, P.; Klingspor, S.; Aschenbach, J.R. Effects of the probiotic Enterococcus faecium and pathogenic Escherichia coli strains in a pig and human epithelial intestinal cell model. Scientifica 2015, 235184. [Google Scholar] [CrossRef]
- Koruri, S.S.; Chowdhury, R.; Bhattacharya, P. Potentiation os functional and antimicrobial activities through synergistic growth of probiotic Pediococcus acidilactici with natural prebiotics (garlic, basil). In Microbes in the Spotlight: Recent Progress in the Understanding of Beneficial and Harmful Microorganisms; Méndez-Vilas, A., Ed.; BrownWalker Press: Boca Raton, FL, USA, 2016; pp. 219–224. [Google Scholar]
- Kandylis, P.; Pissaridi, K.; Bekatorou, A.; Kanellaki, M.; Koutinas, A.A. Dairy and non-dairy probiotic beverages. Curr. Opin. Food Sci. 2016, 7, 58–63. [Google Scholar] [CrossRef]
- Adejuwon, A.O.; Oluduro, A.O.; Agboola, F.K.; Olutiola, P.O.; Burkhardt, B.A.; Segal, S.J. Expression of α-amylase by Aspergillus niger: Effect of nitrogen source of growth medium. Adv. Biosci. Bioeng. 2015, 3, 12–19. [Google Scholar]
- Ploss, T.N.; Reilman, E.; Monteferrante, C.G.; Denham, E.L.; Piersma, S.; Lingner, A.; Vehmaanperä, J.; Lorenz, P.; DijlEmail, J.M. Homogeneity and heterogeneity in amylase production by Bacillus subtilis under different growth conditions. Microb. Cell Fact. 2016, 15, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.M.; Thys, R.C.S.; Rodrigues, R.C. Microbial enzymes as substitutes of chemical additives in baking wheat flour-part II: Combined effects of nine enzymes on dough rheology. Food Bioprocess Tech. 2016, 9, 1598–1611. [Google Scholar] [CrossRef]
- Reginatto, C.; Rossi, C.; Miglioranza, B.G.; Santos, M.; Meneghel, L.; Silveira, M.M.; Malvessi, E. Pectinase production by Aspergillus niger LB-02-SF is influenced by the culture medium composition and the addition of the enzyme inducer after biomass growth. Process Biochem. 2017, 58, 1–8. [Google Scholar] [CrossRef]
- Kalac, P. Edible Mushrooms: Chemical Composition and Nutritional Value; Academic Press: Oxford, UK, 2016; 236p. [Google Scholar]
- Cruz, A.; Pimentel, L.; Rodríguez-Alcalá, R.; Fernandes, T.; Pintado, M. Health benefits of edible mushrooms focused on Coriolus versicolor: A review. J. Food Nutr. Res. 2016, 4, 773–781. [Google Scholar] [CrossRef]
- Chang, R. Functional properties of edible mushrooms. Nutr. Rev. 1996, 54, S91–S93. [Google Scholar] [CrossRef] [PubMed]
- Anupama, R.P. Value-added food: Single cell protein. Biotechnol. Adv. 2000, 18, 459–479. [Google Scholar] [CrossRef]
- Matassa, S.; Boon, N.; Pikaar, I.; Verstraete, W. Microbial protein: Future sustainable food supply route with low environmental footprint. Microb. Biotechnol. 2016, 9, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Nalage, D.N.; Khedkar, G.D.; Kalyankar, A.D.; Sarkate, A.P.; Ghodke, S.R.; Bedre, V.B. Single cell proteins. In The Encyclopedia of Food and Health; Caballero, B., Finglas, P., Toldrá, F., Eds.; Academic Press: Oxford, UK, 2016; pp. 790–794. [Google Scholar] [CrossRef]
- Borghesi, A.; Stronati, M. Superbugs and antibiotics in the newborn. J. Pediatr. Neonat. Individ. Med. 2015, 4, e040253. [Google Scholar] [CrossRef]
- Adrio, J.L.; Demain, A.L. Microbial enzymes: Tools for biotechnological processes. Biomolecules 2014, 4, 117–139. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.S.; Ray, R.C.; Rosell, C.M.; Panda, D. Microbial enzymes in food applications–history of progress. In Microbial Enzyme Technology in Food Applications; Ray, R.C., Rossell, C.M., Eds.; CRC Press: Boca Raton, FL, USA, 2016; pp. 3–18. [Google Scholar]
- Aehle, W. Enzymes in Industry: Production and Applications; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007; 516p. [Google Scholar]
- Panda, S.K.; Mishra, S.S.; Kayitesi, E.; Ray, R.C. Microbial-processing of fruit and vegetable wastes for production of vital enzymes and organic acids: Biotechnology and scopes. Environ. Res. 2016, 146, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Berrio-Restrepo, J.M.; Saldarriaga, J.C.; Correa, M.A.; Aguirre, N.J. Extracellular enzymatic activity of two hydrolases in wastewater treatment for biological nutrient removal. Appl. Microbiol. Biotechnol. 2017, 101, 7385–7396. [Google Scholar] [CrossRef] [PubMed]
- Kreutz, J.A.; Böckenhüser, I.; Wacht, M.; Fischer, K. A 1-year study of the activities of seven hydrolases in a communal wastewater treatment plant: Trends and correlations. Appl. Microbiol. Biotechnol. 2016, 100, 6903–6915. [Google Scholar] [CrossRef] [PubMed]
- Hasan, F.; Shah, A.A.; Hameed, A. Industrial applications of microbial lipases. Enzyme Microb. Technol. 2006, 39, 235–251. [Google Scholar] [CrossRef]
- Jones, E.E.; Bienkowski, D.A.; Stewart, A. The importance of water potential range tolerance as a limiting factor on Trichoderma spp. biocontrol of Sclerotinia sclerotiorum. Ann. Appl. Biol. 2016, 168, 41–51. [Google Scholar] [CrossRef]
- Jones, E.E.; Rabeendran, N.; Stewart, A. Biocontrol of Sclerotinia sclerotiorum infection of cabbage by C. minitans and Trichoderma spp. Biocontrol. Sci. Technol. 2014, 24, 1363–1382. [Google Scholar] [CrossRef]
- Saravanakumar, K.; Yu, C.; Dou, K.; Wang, M.; Li, Y.; Chen, J. Synergistic effect of Trichoderma-derived antifungal metabolites and cell wall degrading enzymes on enhanced biocontrol of Fusarium oxysporum f. sp. cucumerinum. Biol. Control 2016, 94, 37–46. [Google Scholar] [CrossRef]
- Carrero-Carrón, I.; Trapero-Casas, J.L.; Olivares-García, C.; Monte, E.; Hermosa, R.; Jiménez-Díaz, R.M. Trichoderma asperellum is effective for biocontrol of Verticillium wilt in olive caused by the defoliating pathotype of Verticillium dahlia. Crop Prot. 2016, 88, 45–52. [Google Scholar] [CrossRef]
- Khaledi, N.; Taheri, P. Biocontrol mechanisms of Trichoderma harzianum against soybean charcoal rot caused by Macrophomina phaseolina. J. Plant Prot. Res. 2016, 56, 21–31. [Google Scholar] [CrossRef]
- Sokhandani, Z.; Moosavi, M.R.; Basirnia, T. Optimum concentrations of Trichoderma longibrachiatum and cadusafos for controlling Meloidogyne javanica on Zucchini plants. J. Nematol. 2016, 48, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Feyisa, B.; Lencho, A.; Selvaraj, T.; Getaneh, G. Evaluation of some botanicals and Trichoderma harzianum against root-knot nematode (Meloidogyne incognita (Kofoid and White) Chit wood) in tomato. J. Entomol. Nematol. 2016, 8, 11–18. [Google Scholar] [CrossRef]
- Sahebani, N.; Hadavi, N. Biological control of the root-knot nematode Meloidogyne javanica by Trichoderma harzianum. Soil Biol. Biochem. 2008, 40, 2016–2020. [Google Scholar] [CrossRef]
- Kuan, K.B.; Othman, R.; Rahim, K.A.; Shamsuddin, Z.H. Plant growth-promoting rhizobacteria inoculation to enhance vegetative growth, nitrogen fixation and nitrogen remobilisation of maize under greenhouse conditions. PLoS ONE 2016, 11, e0152478. [Google Scholar] [CrossRef] [PubMed]
- Puri, A.; Padda, K.P.; Chanway, C.P. Evidence of nitrogen fixation and growth promotion in canola (Brassica napus L.) by an endophytic diazotroph Paenibacillus polymyxa P2b-2R. Biol. Fert. Soils 2016, 52, 119–125. [Google Scholar] [CrossRef]
- Spaepen, S. Plant hormones produced by microbes. In Principles of Plant-Microbe Interactions; Lugtenberg, B., Ed.; Springer: Cham, Switzerland, 2015; pp. 247–256. [Google Scholar]
- Ahmad, S.; Abbas, S.S.; Prakash, R.; Alam, A.; Husain, M.A. Applications of endophytic Actinomycetes and their role in protection. Imp. J. Interdiscip. Res. 2016, 1–2, 854–858. [Google Scholar]
- Kumar, A.; Bahadur, I.; Maurya, B.R.; Raghuwanshi, R.; Meena, V.S.; Singh, D.K. Does a plant growth promoting rhizobacteria enhance agricultural sustainability? J. Pure Appl. Microbiol. 2015, 9, 715–724. [Google Scholar]
- Awadhiya, A.; Tyeb, S.; Rathore, K.; Verma, V. Agarose bioplastic-based drug delivery system for surgical and wound dressings. Eng. Life Sci. 2016, 17, 204–214. [Google Scholar] [CrossRef]
- Ivanov, V.; Stabnikov, V. Construction biotechnological plastics. In Construction Biotechnology; Ivanov, V., Stabnikov, V., Eds.; Springer: Singapore, 2017; pp. 51–75. [Google Scholar]
- Blackwell, M. The fungi: 1, 2, 3 … 5.1 million species? Am. J. Bot. 2011, 98, 426–438. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitorino, L.C.; Bessa, L.A. Microbial Diversity: The Gap between the Estimated and the Known. Diversity 2018, 10, 46. https://doi.org/10.3390/d10020046
Vitorino LC, Bessa LA. Microbial Diversity: The Gap between the Estimated and the Known. Diversity. 2018; 10(2):46. https://doi.org/10.3390/d10020046
Chicago/Turabian StyleVitorino, Luciana Cristina, and Layara Alexandre Bessa. 2018. "Microbial Diversity: The Gap between the Estimated and the Known" Diversity 10, no. 2: 46. https://doi.org/10.3390/d10020046
APA StyleVitorino, L. C., & Bessa, L. A. (2018). Microbial Diversity: The Gap between the Estimated and the Known. Diversity, 10(2), 46. https://doi.org/10.3390/d10020046