
N,N,N′-Tris(trimethylsilyl)-2-pyridinecarboximidamide



Abstract
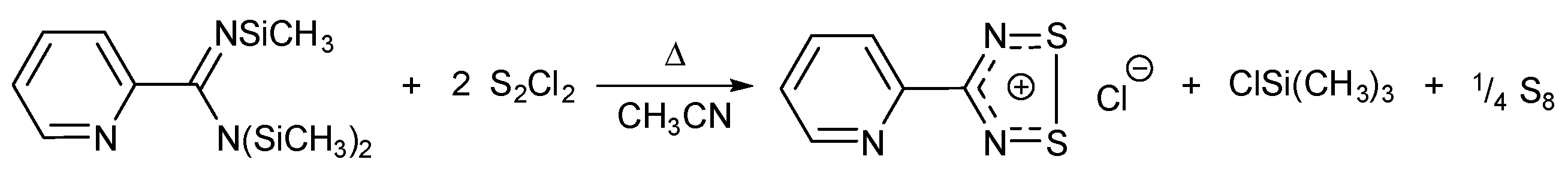
1. Introduction

2. Results and Discussion
2.1. Preparation, Identification and SC-XRD Structure Model
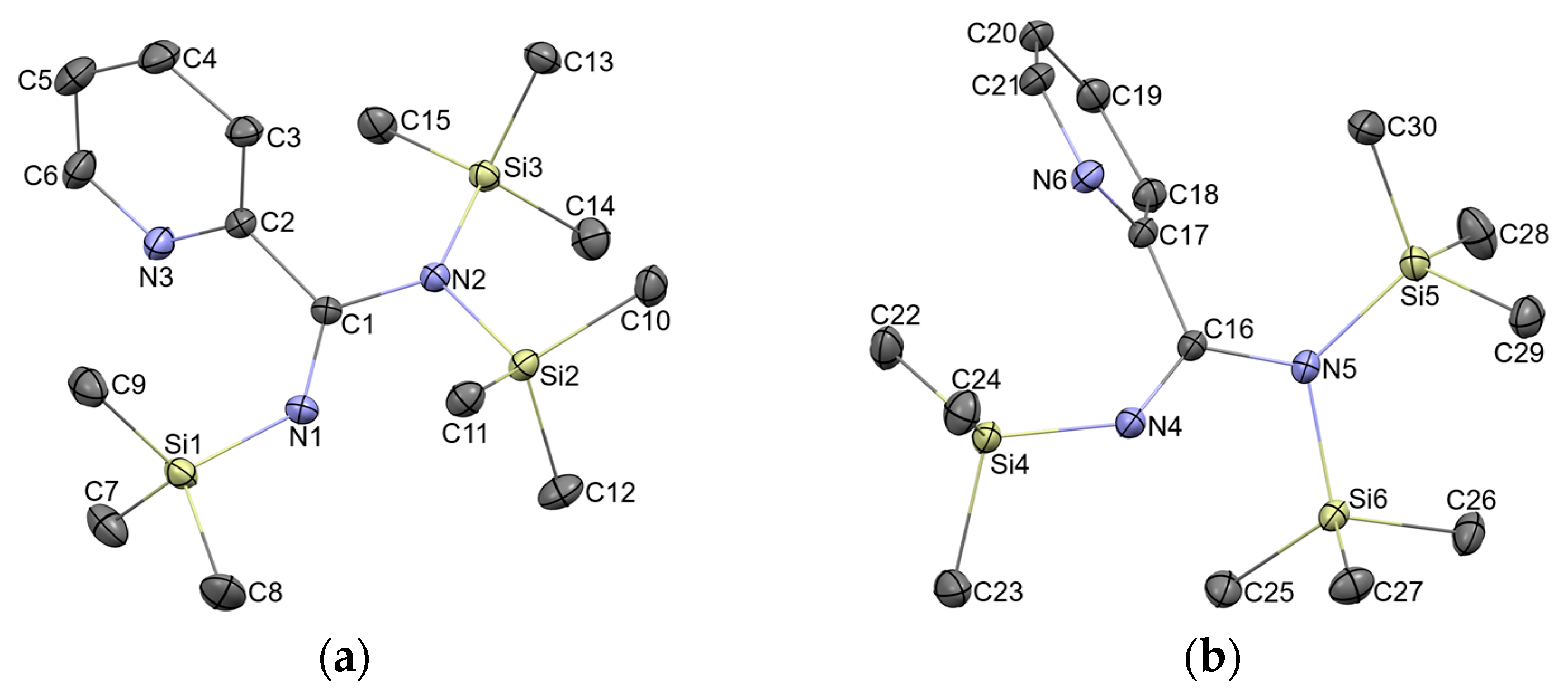
2.2. Molecular Structure Description of 1 as Found in Crystals
2.3. Molecular Structure of 1 Compared to the Literature
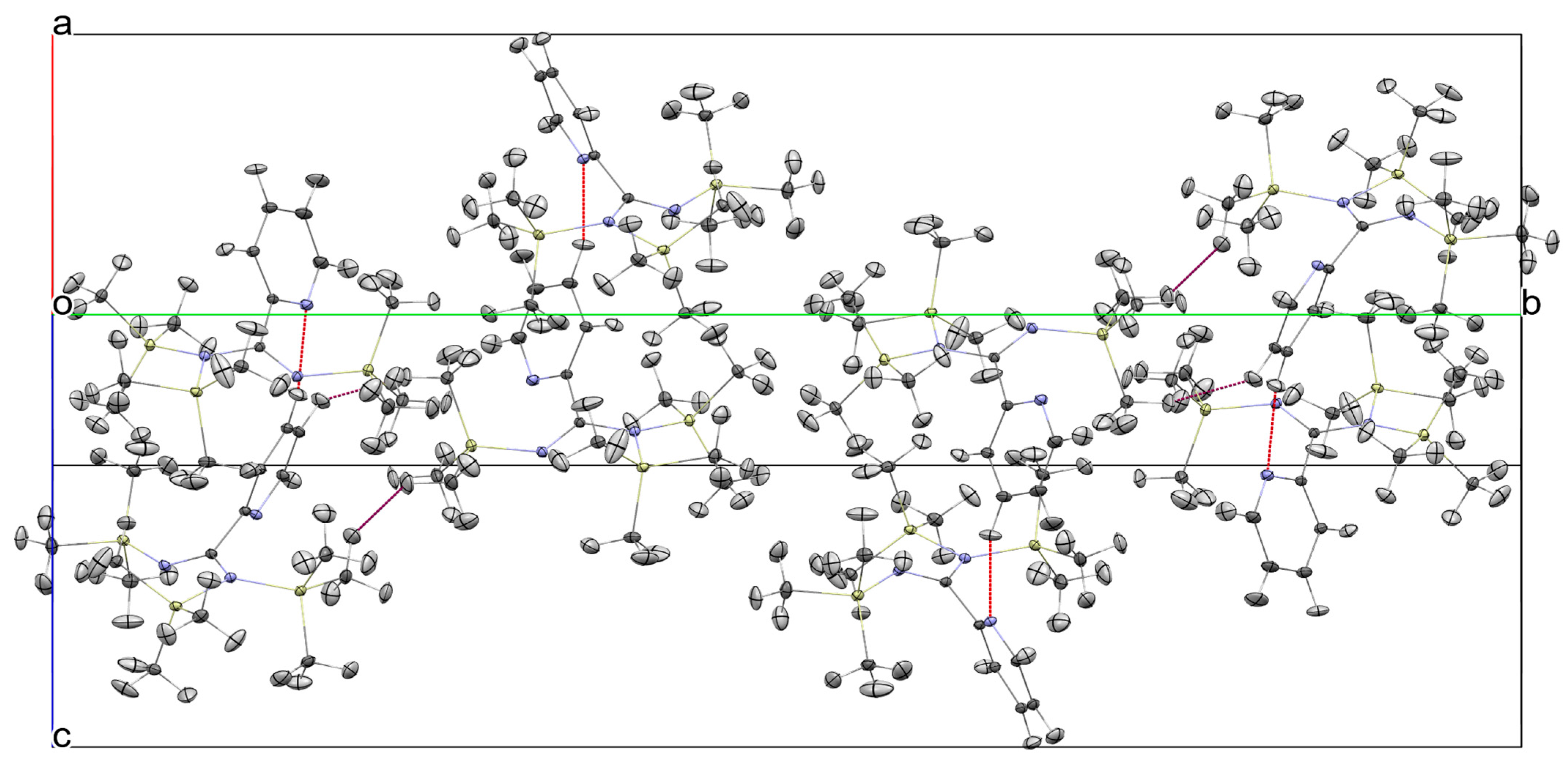
2.4. Lattice Structure of 1
3. Discussion
4. Materials and Methods
General Materials and Procedures
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| Dcalc | Calculated density from SC-XRD |
| DFTX | Density functional theory |
| HAR | Hirshfeld Atom Refinement |
| NMR | Nuclear Magnetic Resonance |
| NoSpherA2 | NOn-SPHERical Atom-form-factors in Olex2 |
| PyDTDA | 4-(2-pyridyl)-1,2-dithia-3,5-diazolyl |
| SC-XRD | Single-Crystal X-ray Diffraction |
| s.u. | standard uncertainty from least squares refinement |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | 1 (C1) | 1 (C16) | 2 | 3 | 4 | 5 | Average |
|---|---|---|---|---|---|---|---|
| C1-N1 | 1.2777(11) | 1.2774(11) | 1.268(3) | 1.279(7) | 1.270(7) | 1.270(3) | 1.274 ± 0.004 |
| C1-N2 | 1.3958(11) | 1.3978(10) | 1.411(3) | 1.403(5) | 1.414(7) | 1.413(3) | 1.406 ± 0.007 |
| C1-C2 | 1.5068(11) | 1.5033(11) | 1.497(2) | 1.503(6) | 1.505(7) | 1.507(4) | 1.504 ± 0.003 |
| N1-Si1 | 1.7362(8) | 1.7340(7) | 1.722(2) | 1.729(4) | 1.715(4) | 1.724(2) | 1.727 ± 0.007 |
| N2-Si2 | 1.7881(7) | 1.7911(7) | 1.775(2) | 1.770(4) | 1.777(5) | 1.777(2) | 1.780 ± 0.007 |
| N2-Si3 | 1.7746(7) | 1.7730(7) | 1.766(2) | 1.774(4) | 1.762(4) | 1.766(3) | 1.769 ± 0.005 |
| N2-C1-N1 | 120.89(8) | 120.71(7) | 119.6(2) | 119.8(4) | 120.2(5) | 120.4(2) | 120.3 ± 0.5 |
| C2-C1-N1 | 121.77(8) | 121.35(7) | 123.5(2) | 123.6(4) | 124.5(5) | 123.5(2) | 123.0 ± 1.1 |
| C2-C1-N2 | 117.16(7) | 117.75(7) | 116.6(2) | 116.6(4) | 115.1(4) | 115.9(2) | 116.5 ± 0.9 |
| C1-N1-Si1 | 132.65(6) | 134.69(7) | 136.6(2) | 136.6(3) | 138.7(4) | 139.0(2) | 136.4 ± 2.2 |
| C1-N2-Si2 | 112.69(5) | 111.72(6) | 111.9(1) | 113.1(3) | 112.2(4) | 113.3(2) | 112.5 ± 0.6 |
| C1-N2-Si3 | 123.88(6) | 124.10(6) | 123.1(1) | 122.1(3) | 121.7(4) | 121.4(2) | 122.7 ± 1.0 |
| Si2-N2-Si3 | 122.35(4) | 122.80(4) | 124.5(1) | 123.1(2) | 125.0(3) | 124.2(1) | 123.7 ± 1.0 |
| ∠(arom/amid) 2 | 51.86 | 54.67 | 56.13 | 48.08 | 48.73 | 51.40 | 51.4 ± 3.1 |
References
- Boeré, R.T.; Oakley, R.T.; Reed, R.W. Preparation of N,N,N′-tris(trimethylsilyl)amidines. A convenient route to unsubstituted amidines. J. Organomet. Chem. 1987, 331, 161–167. [Google Scholar] [CrossRef]
- Boeré, R.T.; Hicks, R.G.; Oakley, R.T. N,N,N′-Tris(trimethylsilyl)amidines. In Inorganic Syntheses; Wiley: Hoboken, NJ, USA, 1997; Volume 31, pp. 94–98. ISBN 978-0-470-13297-5. [Google Scholar]
- Hearns, N.G.R.; Preuss, K.E.; Richardson, J.F.; Bin-Salamon, S. Design and synthesis of a 4-(2′-pyridyl)-1,2,3,5-dithiadiazolyl cobalt complex. J. Am. Chem. Soc. 2004, 126, 9942–9943. [Google Scholar] [CrossRef] [PubMed]
- Del Bel Belluz, P.; Cordes, A.W.; Kristof, E.M.; Kristof, P.V.; Liblong, S.W.; Oakley, R.T. 1,2,3,5-Diselenadiazolyls as building blocks for molecular metals. Preparation and structures of [PhCN2Se2]+PF6- and [PhCN2Se2]2. J. Am. Chem. Soc. 1989, 111, 9276–9278. [Google Scholar] [CrossRef]
- Boeré, R.T.; Hicks, R.G.; Oakley, R.T. Synthesis and interconversion of 5-phenyl-1,3,2,4,6-dithiatriazine derivatives; crystal and molecular structure of the bicyclic molecule PhCN5S3. J. Chem. Soc. Chem. Commun. 1985, 929–930. [Google Scholar] [CrossRef]
- Chivers, T.; Parvez, M.; Vargas-Baca, I.; Ziegler, T.; Zoricak, P. Formation and X-ray Structures of Eight- and Sixteen-Membered Rings (ArC)nN2n(SPh)n [n = 2, Ar = 4-XC6H4 (X = Br, CF3); n = 4, Ar = 4-BrC6H4] and the Electronic Structures of (HC)2N4(SH)2 and (HC)2N4(SH)22. Inorg. Chem. 1997, 36, 1669–1675. [Google Scholar] [CrossRef]
- Edelmann, F. N-silylated benzamidines: Versatile building blocks in main group and coordination chemistry. Coord. Chem. Rev. 1994, 137, 403–481. [Google Scholar] [CrossRef]
- Edelmann, F. Chapter 3—Advances in the Coordination Chemistry of Amidinate and Guanidinate Ligands. Adv. Organomet. Chem. 2008, 57, 183–352. [Google Scholar] [CrossRef]
- Britten, J.; Hearns, N.G.R.; Preuss, K.E.; Richardson, J.F.; Bin-Salamon, S. Mn(II) and Cu(II) Complexes of a Dithiadiazolyl Radical Ligand: Monomer/Dimer Equilibria in Solution. Inorg. Chem. 2007, 46, 3934–3945. [Google Scholar] [CrossRef]
- Hearns, N.G.R.; Fatila, E.M.; Clérac, R.; Jennings, M.; Preuss, K.E. Ni(II) and hs-Fe(II) Complexes of a Paramagnetic Thiazyl Ligand, and Decomposition Products of the Iron Complex, Including an Fe(III) Tetramer. Inorg. Chem. 2008, 47, 10330–10341. [Google Scholar] [CrossRef]
- Hearns, N.G.R.; Clérac, R.; Jennings, M.; Preuss, K.E. Manipulating the crystal packing of pyDTDA radical ligand coordination complexes with Mn(II) and Ni(II). Dalton Trans. 2009, 3193–3203. [Google Scholar] [CrossRef] [PubMed]
- Fatila, E.M.; Mayo, R.A.; Rouzières, M.; Jennings, M.C.; Dechambenoit, P.; Soldatov, D.V.; Mathonière, C.; Clérac, R.; Coulon, C.; Preuss, K.E. Radical−Radical Recognition: Switchable Magnetic Properties and Re-entrant Behavior. Chem. Mater. 2015, 27, 4023–4032. [Google Scholar] [CrossRef]
- Mills, M.B.; Hollingshead, A.G.; Maahs, A.C.; Soldatov, D.V.; Preuss, K.E. Isomerization of a lanthanide complex using a humming top guest template: A solid-to-solid reaction. CrystEngComm 2015, 17, 7816–7819. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef]
- Kleemiss, F.; Dolomanov, O.V.; Bodensteiner, M.; Peyerimhoff, N.; Midgley, L.; Bourhis, L.J.; Genoni, A.; Malaspina, L.A.; Jayatilaka, D.; Spencer, J.L.; et al. Accurate crystal structures and chemical properties from NoSpherA2. Chem. Sci. 2021, 12, 1675–1692. [Google Scholar] [CrossRef]
- Ergezinger, C.; Weller, F.; Dehnicke, K.Z. Die Kristallstruktur von N,N,N′-tris(trimethylsilyl)benzamidin sowie die Synthese und Kristallstruktur von Dichlorantimon-N,N′-bis(trimethylsilyl)benzamidinat. Naturforsch. B Chem. Sci. 1988, 43, 1119–1124. [Google Scholar] [CrossRef]
- Lu, Z.; Hill, N.J.; Findlater, M.; Cowley, A.H. Diboron complexes of binucleating bis(amidinate) ligands. Inorg. Chim. Acta 2007, 360, 1316–1322. [Google Scholar] [CrossRef]
- Beekman, R.A.; Boeré, R.T.; Moock, K.H.; Parvez, M. Synthesis, electrochemistry, structure, and magnetic susceptibility of 5-tert-butyl-1,3-bis-(1,2,3,5-dithiadiazolyl)benzene. Structural effect of the bulky substituent. Can. J. Chem. 1998, 76, 85–93. [Google Scholar] [CrossRef]
- Weller, F.; Schmock, F.; Dehnicke, K. Synthese und Kristallstruktur von 2-Trimethylsilyl-1,3-bistrimethylsilylimino-1,3-dihydro-isoindol sowie die Kristallstruktur von Hexakis(trimethylsilyl)-1,4-benzdiamidin. Z. Naturforsch. B Chem. Sci. 1989, 44, 548–552. [Google Scholar] [CrossRef]
- Hill, N.D.D.; Boeré, R.T. Small Molecule X-ray Crystal Structures at a Crossroads. Chemistry—Methods 2025, 5, e202400052. [Google Scholar] [CrossRef]
- Häfelinger, G.; Kuske, K.H. The Chemistry of the Amidines and Imidates; Patai, S., Rappoport, Z., Eds.; Wiley: Chichester, UK, 1991; Volume 2, Chapter 1; pp. 1–100. [Google Scholar]
- Marszaukowski, F.; Boeré, R.T.; Wohnrath, K. Frustrated and Realized Hydrogen Bonding in 4-Hydroxy-3,5-ditertbutylphenylphosphine Derivatives. Cryst. Growth Des. 2022, 22, 2512–2533. [Google Scholar] [CrossRef]
- Allen, F.H.; Bruno, I.J. Bond lengths in organic and metal-organic compounds revisited: X─H bond lengths from neutron diffraction data. Acta Cryst. B 2010, 66, 380–386. [Google Scholar] [CrossRef]
- Knapp, C.; Lork, E.; Watson, P.G.; Mews, R. Lithium Fluoroarylamidinates: Syntheses, Structures, and Reactions. Inorg. Chem. 2002, 41, 2014–2025. [Google Scholar] [CrossRef] [PubMed]
- Lisovskii, A.; Botoshanskya, M.; Eisen, M.S. Towards the formation of dimeric and trimeric lithium benzamidinates: Characterization of intermediate structures. J. Chem. Soc. Dalton Trans. 2001, 11, 1692–1698. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Boeré, R.T. Hydrogen Bonds Stabilize Chloroselenite Anions: Crystal Structure of a New Salt and Donor-Acceptor Bonding to SeO2. Molecules 2023, 28, 7489. [Google Scholar] [CrossRef] [PubMed]
- Aharonovich, S.; Kapon, M.; Botoshanski, M.; Eisen, M.S. N,N′-Bis-Silylated Lithium Aryl Amidinates: Synthesis, Characterization, and the Gradual Transition of Coordination Mode from σ Toward π Originated by Crystal Packing Interactions. Organometallics 2008, 27, 1869–1877. [Google Scholar] [CrossRef]
- Rabinovich, E.; Aharonovich, S.; Botoshanski, M.; Eisen, M.S. Thorium 2-pyridylamidinates: Synthesis, structure and catalytic activity towards the cyclo-oligomerization of ε-caprolactone. Dalton Trans. 2010, 39, 6667–6676. [Google Scholar] [CrossRef]
- Domeshek, E.; Batrice, R.J.; Aharonovich, S.; Tumanskiii, B.; Botoshanski, M.; Eisen, M.S. Organoactinides in the polymerization of ethylene: Is TIBA a better cocatalyst than MAO? Dalton Trans. 2013, 42, 9069–9078. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H.J. OLEX2: A complete structure solution, refinement and analysis program. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment—Olex2 dissected. Acta Cryst. 2015, A71, 59–75. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system—Version 5.0. WIREs Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
| Parameter | Molecule C1 | Molecule C16 | Av. Compar. 2 |
|---|---|---|---|
| C1-N1 | 1.2777(11) | 1.2774(11) | 1.274 ± 0.004 |
| C1-N2 | 1.3958(11) | 1.3978(10) | 1.406 ± 0.007 |
| C1-C2 | 1.5068(11) | 1.5033(11) | 1.504 ± 0.003 |
| N1-Si1 | 1.7362(8) | 1.7340(7) | 1.727 ± 0.007 |
| N2-Si2 | 1.7881(7) | 1.7911(7) | 1.780 ± 0.007 |
| N2-Si3 | 1.7746(7) | 1.7730(7) | 1.769 ± 0.005 |
| C2-N3 | 1.3408(11) | 1.3409(11) | n.a. |
| C2-C3 | 1.3957(12) | 1.3955(12) | n.a. |
| C3-C4 | 1.3919(14) | 1.3920(13) | n.a. |
| C4-C5 | 1.3933(16) | 1.3923(14) | n.a. |
| C5-C6 | 1.3929(15) | 1.3918(14) | n.a. |
| C6-N3 | 1.3388(12) | 1.3368(11) | n.a. |
| N2-C1-N1 | 120.89(8) | 120.71(7) | 120.3 ± 0.5 |
| C2-C1-N1 | 121.77(8) | 121.34(7) | 123.0 ± 1.1 |
| C2-C1-N2 | 117.16(7) | 117.75(7) | 116.5 ± 0.9 |
| C1-N1-Si1 | 132.65(6) | 134.69(6) | 136.4 ± 2.2 |
| C1-N2-Si2 | 112.69(5) | 111.72(6) | 112.5 ± 0.6 |
| C1-N2-Si3 | 123.88(6) | 124.10(6) | 122.7 ± 1.0 |
| Si2-N2-Si3 | 122.35(4) | 122.80(4) | 123.7 ± 1.0 |
| ∠(arom/amid) 3 | 51.86(5) | 54.67(5) | 51.4 ± 3.1 |
| D | H | A | d(D–H)/Å | d(H∙∙∙A)/Å | d(D∙∙∙A)/Å | D–H∙∙∙A/° |
|---|---|---|---|---|---|---|
| C4 | H4 | N6 | 1.110(12) | 2.390(12) | 3.4649(13) | 162.4(10) |
| C19 | H19 | N3 1 | 1.089(12) | 2.435(12) | 3.4840(13) | 161.2(10) |
| H9C | H20 2 | 2.165(18) | 154.8(14) | |||
| H8C | H30A 2 | 2.180(20) | 132.9(13) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, M.A.; Preuss, K.E.; Boeré, R.T. N,N,N′-Tris(trimethylsilyl)-2-pyridinecarboximidamide. Molbank 2025, 2025, M2031. https://doi.org/10.3390/M2031
Ibrahim MA, Preuss KE, Boeré RT. N,N,N′-Tris(trimethylsilyl)-2-pyridinecarboximidamide. Molbank. 2025; 2025(3):M2031. https://doi.org/10.3390/M2031
Chicago/Turabian StyleIbrahim, Mukaila A., Kathryn E. Preuss, and René T. Boeré. 2025. "N,N,N′-Tris(trimethylsilyl)-2-pyridinecarboximidamide" Molbank 2025, no. 3: M2031. https://doi.org/10.3390/M2031
APA StyleIbrahim, M. A., Preuss, K. E., & Boeré, R. T. (2025). N,N,N′-Tris(trimethylsilyl)-2-pyridinecarboximidamide. Molbank, 2025(3), M2031. https://doi.org/10.3390/M2031