
5,5′-Di((E)-buta-1,3-dien-1-yl)-2,2′,3,3′-tetramethoxy-1,1′-biphenyl



Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
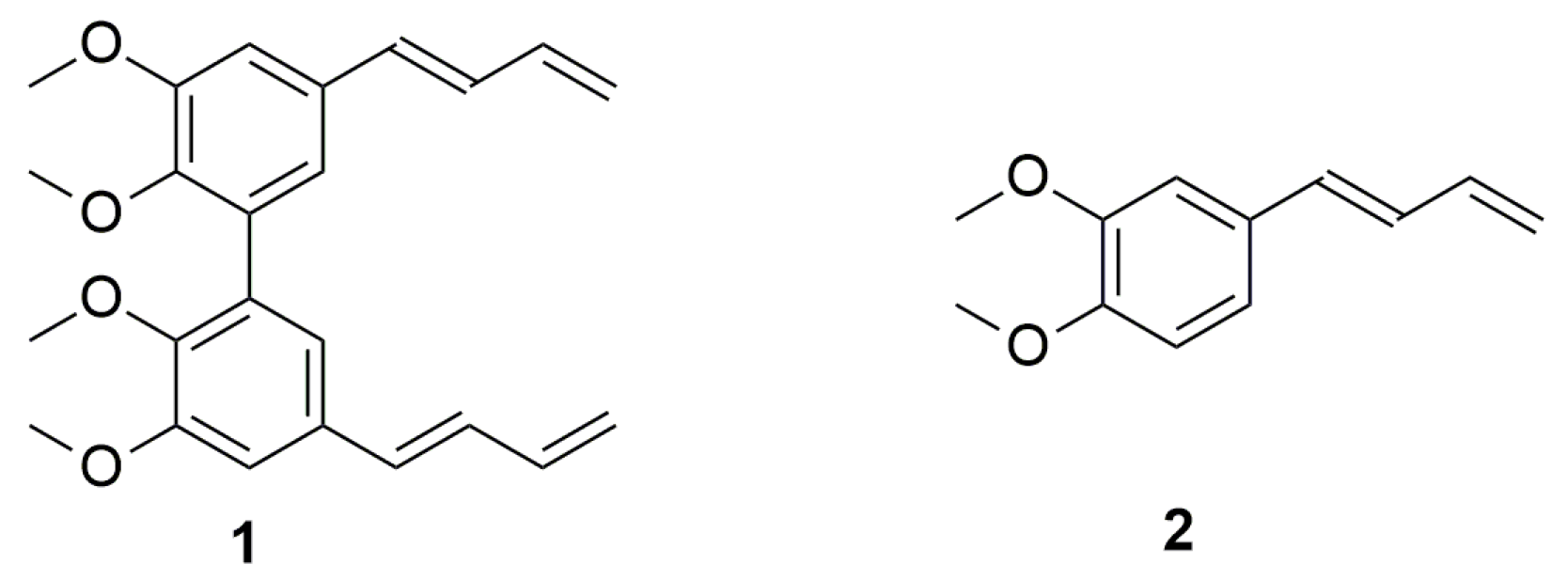
1. Introduction
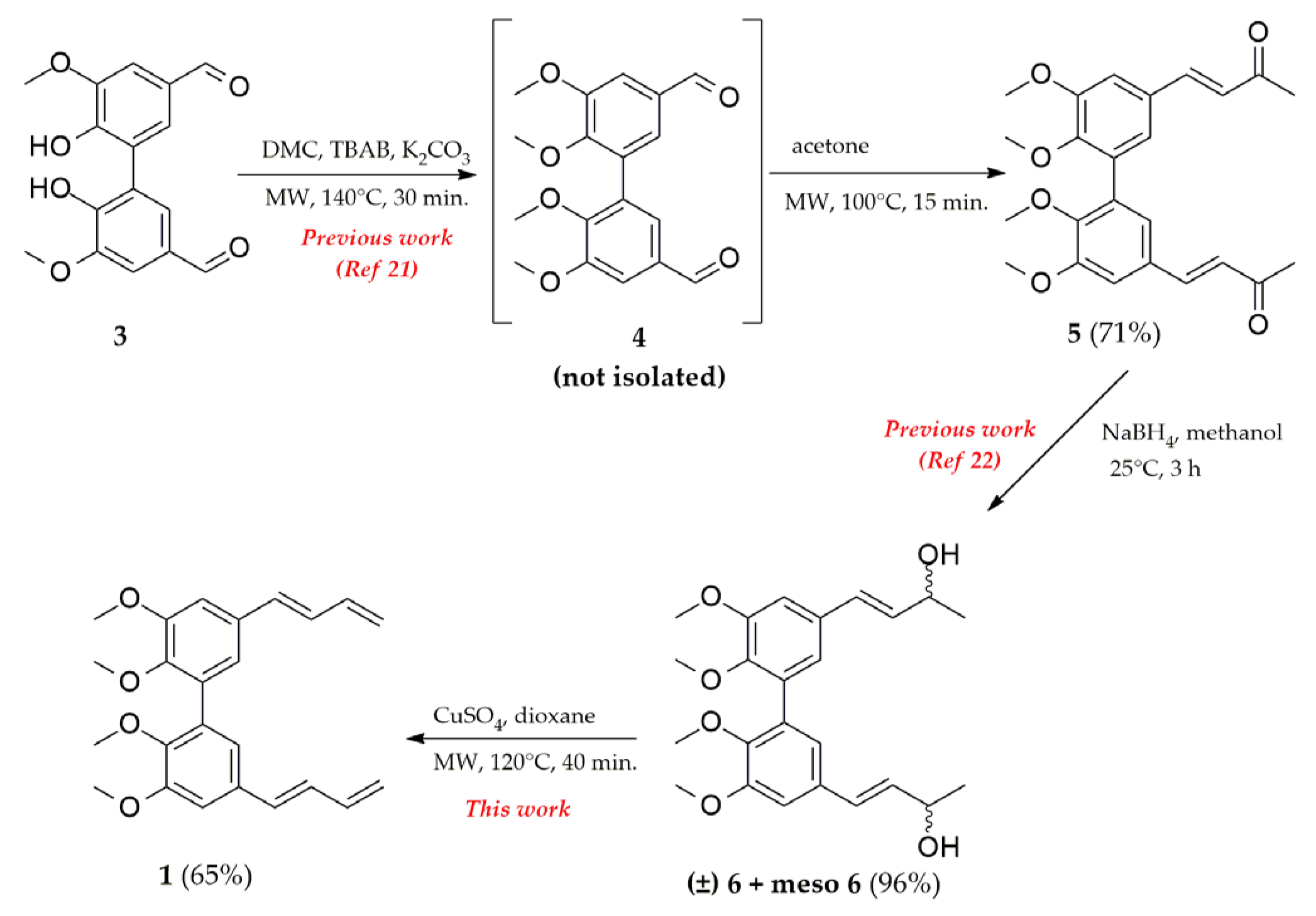
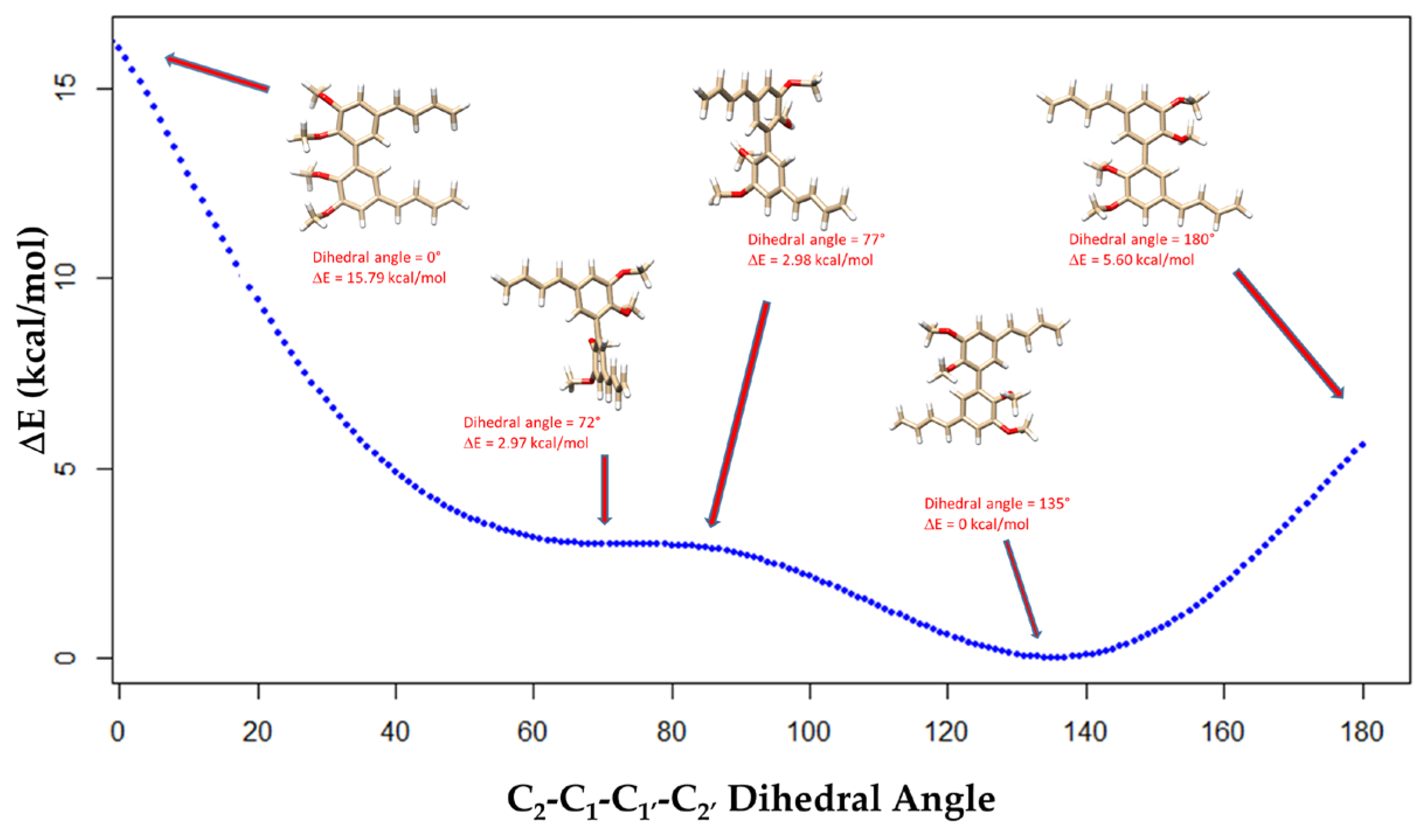
2. Results and Discussion
3. Materials and Methods
- Synthesis of 1,1′-(5,5′,6,6′-tetramethoxy-[1,1′-biphenyl]-3,3′-diyl)bis(but-3-en-1-ol) 7.
- To a solution of 4 (0.5 g, 1.5 mmol) in tetrahydrofuran (5 mL), allyl bromide (0.93 mL, 10.7 mmol) and saturated aqueous ammonium chloride solution (10 mL) were added. The reaction mixture was cooled to 0° C. Zinc powder (1.4 g, 21.4 mmol) was added, and the solution was stirred at 0 °C for 30 min. The resulting precipitate was filtered, and the filtrate was extracted with ethyl acetate. The organic layers were combined and washed with brine, dried over sodium sulfate, filtered, and concentrated in vacuo. The crude compound was purified by flash chromatography using a 1:2 mixture of ethyl acetate/petroleum ether as the eluent to obtain 7 as a colorless oil (0.51 g, 90%). 1H-NMR (methanol-d4) δ 2.25 (m, 4H), 3.58 (s, -OCH3, 6H), 3.91 (s, -OCH3, 6H), 4.66 (t, J = 7.2 Hz, 2H), 5.05 (dd, J = 0.6, 9.0 Hz, Ar, 2H), 5.08 (dd, J = 0.6, 16.8 Hz, 2H), 5.82 (m, 2H), 6.79 (d, J = 2.4 Hz, Ar, 2H), 7.05 (d, J = 2.4 Hz, Ar, 2H); 13C-NMR (methanol-d4) δ 43.93, 54.97, 59.60, 73.29, 109.43, 116.11, 120.39, 132.37, 134.72, 140.02, 145.60, 152.52; Anal. Calcd for C24H30O6: C, 69.54; H, 7.30. Found: C, 69.60; H, 7.35; ESI-MS m/z (ES+) Found: [M+Na]+ 437.1941: C24H30O6 Na, requires [M+Na]+ 437.1939.
- Synthesis of 5,5′-di((E)-buta-1,3-dien-1-yl)-2,2′,3,3′-tetramethoxy-1,1′-biphenyl 1.
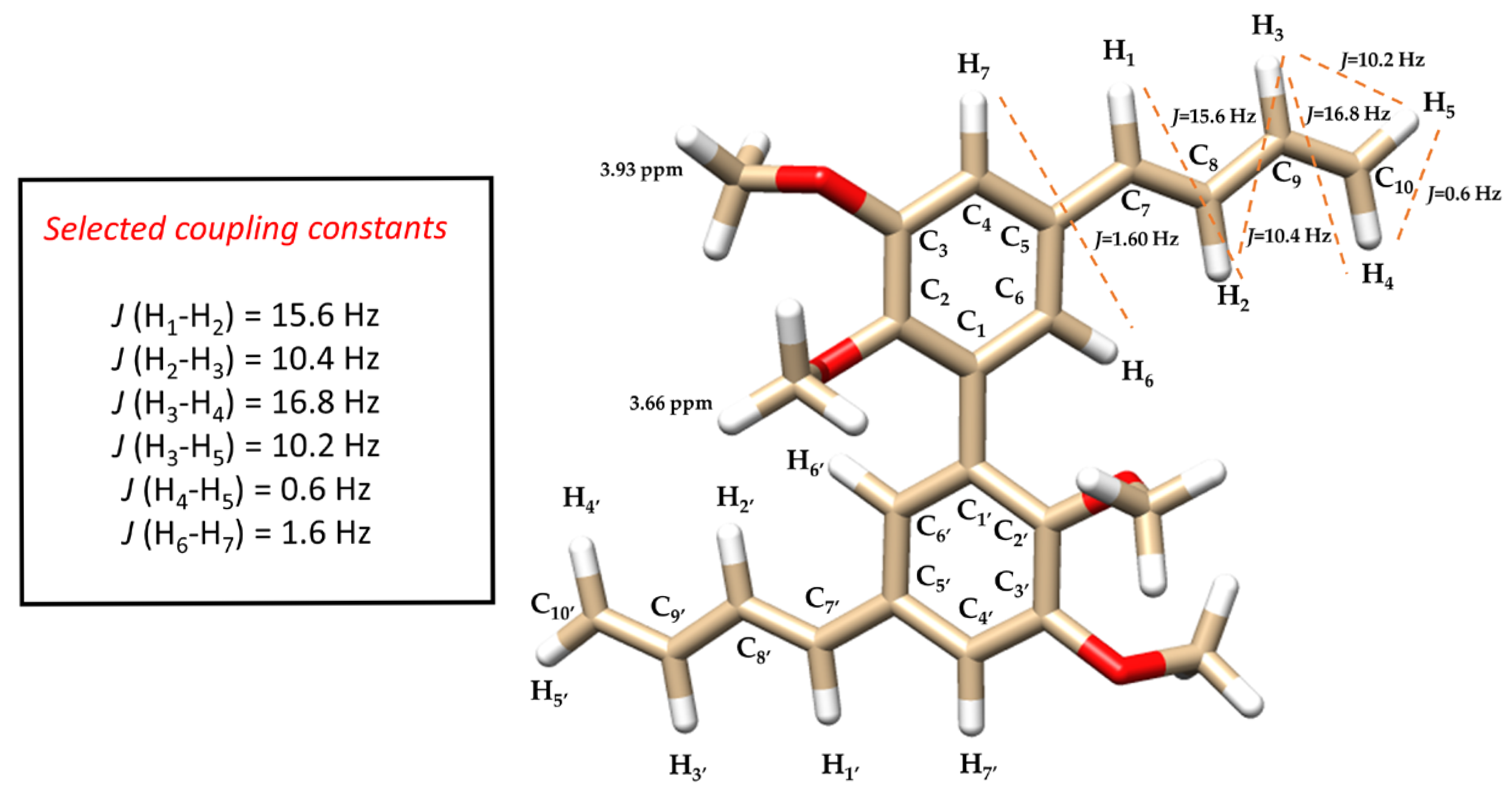
- To a solution of compound 6 or 7 (0.25 g, 0.6 mmol) in dioxane (10 mL), copper sulfate (0.2 g, 1.2 mmol) was added at room temperature. The solution was then stirred under MW irradiation at 120 °C for 40 min. The solution was cooled at rt and filtered. The filtrate was extracted with ethyl acetate. The organic layers were combined and washed with brine, dried over sodium sulfate, filtered, and concentrated in vacuo. The crude material was purified by flash chromatography using a 1:5 mixture of acetone/petroleum ether as the eluent to obtain 1 as a yellow oil (0.15 g, 65% or 0.22 g, 95% starting from diol 6 or 7 respectively). 1H-NMR (methanol-d4) δ 3.66 (s, -OCH3, 6H), 3.93 (s, -OCH3, 6H), 5.14 (dd, J = 0.6, 10.2 Hz, H5-H5′, 2H), 5.32 (dd, J = 0.6, 16.8 Hz, H4-H4′, 2H), 6.47 (m, H3-H3′, 2H), 6.54 (d, J = 15.6 Hz, H1-H1′, 2H), 6.80 (dd, J = 10.4, 15.6 Hz, H2-H2′, 2H), 6.88 (d, J = 1.6 Hz, Ar, 2H), 7.12 (d, J = 1.6 Hz, Ar, 2H); 13C-NMR (methanol-d4) δ 55.00, 59.68, 109.22, 116.15, 121.33, 129.96, 132.19, 132.63, 132.95, 137.21, 146.36, 152.79; Anal. Calcd for C24H26O4: C, 76.17; H, 6.92. Found: C, 76.23; H, 6.95; ESI-MS m/z (ES+) Found: [M+Na]+ 401.1732: C24H26O4 Na, requires [M+Na]+ 401.1728.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seaho, B.; Lekwongphaiboon, C.; Tayana, N.; Inthakusol, W.; Kongkiatpaiboon, S.; Mahavorasirikul, W.; Prateeptongkum, S.; Duangdee, N. Absolute Quantification of Phenylbutanoids in Zingiber cassumunar Roxb. Rhizome by Quantitative1 H NMR. Phytochem. Anal. 2024, 36, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Iwami, J.; Pongpiriyadacha, Y.; Nakashima, S.; Matsuda, H.; Yoshikawa, M. Chemical Structures of Phenylbutanoids From Rhizomes of Zingiber cassumunar. Nat. Prod. Commun. 2022, 17, 1934578X2210778. [Google Scholar] [CrossRef]
- Soberón, J.R.; Sgariglia, M.A.; Sampietro, D.A.; Quiroga, E.N.; Vattuone, M.A. Free Radical Scavenging Activities and Inhibition of Inflammatory Enzymes of Phenolics Isolated from Tripodanthus Acutifolius. J. Ethnopharmacol. 2010, 130, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Panichayupakaranant, P.; Kaewchoothong, A. Preparation of Phenylbutanoid-Rich Zingiber Cassumunar Extracts and Simultaneous HPLC Analysis of Phenylbutanoids. Planta Med. 2012, 78, PI157. [Google Scholar] [CrossRef]
- Kongsui, R.; Sriraksa, N.; Thongrong, S. The Neuroprotective Effect of Zingiber Cassumunar Roxb. Extract on LPS-Induced Neuronal Cell Loss and Astroglial Activation within the Hippocampus. BioMed Res. Int. 2020, 2020, 4259316. [Google Scholar] [CrossRef]
- Sinha, A.K.; Sharma, A.; Joshi, B.P.; Singh, N.P. A Mild Conversion of Phenylpropanoid into Rare Phenylbutanoids: (E)-4-(2′,4′,5′-Trimethoxyphenyl)but-1, 3-Diene and (E)-4-(2′,4′,5′-Trimethoxyphenyl)but-1-Ene Occurring in Zingiber Cassumunar. Nat. Prod. Res. 2005, 19, 771–776. [Google Scholar] [CrossRef]
- Devkota, H.P.; Paudel, K.R.; Hassan, M.M.; Dirar, A.I.; Das, N.; Adhikari-Devkota, A.; Echeverría, J.; Logesh, R.; Jha, N.K.; Singh, S.K.; et al. Bioactive Compounds from Zingiber Montanum and Their Pharmacological Activities with Focus on Zerumbone. Appl. Sci. 2021, 11, 10205. [Google Scholar] [CrossRef]
- Han, A.-R.; Kim, H.; Piao, D.; Jung, C.-H.; Seo, E.K. Phytochemicals and Bioactivities of Zingiber Cassumunar Roxb. Molecules 2021, 26, 2377. [Google Scholar] [CrossRef]
- Li, M.-X.; Ma, Y.-P.; Zhang, H.-X.; Sun, H.-Z.; Su, H.-H.; Pei, S.-J.; Du, Z.-Z. Repellent, Larvicidal and Adulticidal Activities of Essential Oil from Dai Medicinal Plant Zingiber Cassumunar against Aedes Albopictus. Plant Divers. 2021, 43, 317–323. [Google Scholar] [CrossRef]
- Jeenapongsa, R.; Yoovathaworn, K.; Sriwatanakul, K.M.; Pongprayoon, U.; Sriwatanakul, K. Anti-Inflammatory Activity of (E)-1-(3,4-Dimethoxyphenyl) Butadiene from Zingiber Cassumunar Roxb. J. Ethnopharmacol. 2003, 87, 143–148. [Google Scholar] [CrossRef]
- Truong, V.-L.; Manochai, B.; Pham, T.-T.; Jeong, W.-S. Antioxidant and Anti-Inflammatory Activities of Zingiber Montanum Oil in HepG2 Cells and Lipopolysaccharide-Stimulated RAW 264.7 Macrophages. J. Med. Food 2021, 24, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.S. Lignans, Neolignans and Related Compounds. Nat. Prod. Rep. 1999, 16, 75–96. [Google Scholar] [CrossRef]
- Ismail, T.; Calcabrini, C.; Diaz, A.; Fimognari, C.; Turrini, E.; Catanzaro, E.; Akhtar, S.; Sestili, P. Ellagitannins in Cancer Chemoprevention and Therapy. Toxins 2016, 8, 151. [Google Scholar] [CrossRef] [PubMed]
- Saberi Riseh, R.; Fathi, F.; Lagzian, A.; Vatankhah, M.; Kennedy, J.F. Modifying Lignin: A Promising Strategy for Plant Disease Control. Int. J. Biol. Macromol. 2024, 271, 132696. [Google Scholar] [CrossRef]
- Paquin, A.; Reyes-Moreno, C.; Bérubé, G. Recent Advances in the Use of the Dimerization Strategy as a Means to Increase the Biological Potential of Natural or Synthetic Molecules. Molecules 2021, 26, 2340. [Google Scholar] [CrossRef]
- Hajduk, P.J.; Bures, M.; Praestgaard, J.; Fesik, S.W. Privileged Molecules for Protein Binding Identified from NMR-Based Screening. J. Med. Chem. 2000, 43, 3443–3447. [Google Scholar] [CrossRef]
- Heeb, J.-P.; Clayden, J.; Smith, M.D.; Armstrong, R.J. Interrogating the Configurational Stability of Atropisomers. Nat. Protoc. 2023, 18, 2745–2771. [Google Scholar] [CrossRef]
- Kostić, K.; Brborić, J.; Delogu, G.; Simić, M.R.; Samardžić, S.; Maksimović, Z.; Dettori, M.A.; Fabbri, D.; Kotur-Stevuljević, J.; Saso, L. Antioxidant Activity of Natural Phenols and Derived Hydroxylated Biphenyls. Molecules 2023, 28, 2646. [Google Scholar] [CrossRef]
- Setzu, M.D.; Mocci, I.; Fabbri, D.; Carta, P.; Muroni, P.; Diana, A.; Dettori, M.A.; Casu, M.A. Neuroprotective Effects of the Nutraceutical Dehydrozingerone and Its C2-Symmetric Dimer in a Drosophila Model of Parkinson’s Disease. Biomolecules 2024, 14, 273. [Google Scholar] [CrossRef]
- Hodgson, D.M.; Persaud, R.S.D. Convergent Synthesis of Conjugated 1,2-Disubstituted E-Allylic Alcohols from Two Aldehydes and Methylenetriphenylphosphorane. Org. Biomol. Chem. 2012, 10, 7949. [Google Scholar] [CrossRef]
- Dettori, M.A.; Carta, P.; Fabbri, D. Environmentally Friendly One-Pot Two-Step Sequential Synthesis of Biological Active Curcumin Analogues. Tetrahedron 2024, 153, 133867. [Google Scholar] [CrossRef]
- Sanfilippo, C.; Patti, A.; Dettori, M.A.; Fabbri, D.; Delogu, G. Lipase Behavior in the Stereoselective Transesterification of Zingerol-like Derivatives and Related Biphenyls. J. Mol. Catal. B Enzym. 2013, 90, 107–113. [Google Scholar] [CrossRef]
- Kumar, A.; Kuang, Y.; Liang, Z.; Sun, X. Microwave Chemistry, Recent Advancements, and Eco-Friendly Microwave-Assisted Synthesis of Nanoarchitectures and Their Applications: A Review. Mater. Today Nano 2020, 11, 100076. [Google Scholar] [CrossRef]
- Pisano, M.; Pagnan, G.; Dettori, M.A.; Cossu, S.; Caffa, I.; Sassu, I.; Emionite, L.; Fabbri, D.; Cilli, M.; Pastorino, F.; et al. Enhanced Anti-Tumor Activity of a New Curcumin-Related Compound against Melanoma and Neuroblastoma Cells. Mol. Cancer 2010, 9, 137. [Google Scholar] [CrossRef]
- Woodman, P.R.; Munslow, I.J.; Hitchcock, P.B.; Scott, P. Non-Planar Co-Ordination of C2-Symmetric Biaryl-Bridged Schiff-Base Ligands: Well Expressed Chiral Ligand Environments for Zirconium. J. Chem. Soc. Dalton Trans. 1999, 4069–4076. [Google Scholar] [CrossRef]
- Gutiérrez Sanfeliciano, S.M.; Schaus, J.M. Rapid Assessment of Conformational Preferences in Biaryl and Aryl Carbonyl Fragments. PLoS ONE 2018, 13, e0192974. [Google Scholar] [CrossRef]
- Dahlgren, M.K.; Schyman, P.; Tirado-Rives, J.; Jorgensen, W.L. Characterization of Biaryl Torsional Energetics and Its Treatment in OPLS All-Atom Force Fields. J. Chem. Inf. Model. 2013, 53, 1191–1199. [Google Scholar] [CrossRef]
- Mazzanti, A.; Lunazzi, L.; Minzoni, M.; Anderson, J.E. Rotation in Biphenyls with a Single Ortho-Substituent. J. Org. Chem. 2006, 71, 5474–5481. [Google Scholar] [CrossRef]
- Habeck, M.; Rieping, W.; Nilges, M. Bayesian Estimation of Karplus Parameters and Torsion Angles from Three-Bond Scalar Couplings Constants. J. Magn. Reson. 2005, 177, 160–165. [Google Scholar] [CrossRef]
- Charry, J.; Tkatchenko, A. Van Der Waals Radii of Free and Bonded Atoms from Hydrogen (Z = 1) to Oganesson (Z = 118). J. Chem. Theory Comput. 2024, 20, 7469–7478. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dettori, M.A.; Fabbri, D.; Dallocchio, R.; Carta, P. 5,5′-Di((E)-buta-1,3-dien-1-yl)-2,2′,3,3′-tetramethoxy-1,1′-biphenyl. Molbank 2025, 2025, M2018. https://doi.org/10.3390/M2018
Dettori MA, Fabbri D, Dallocchio R, Carta P. 5,5′-Di((E)-buta-1,3-dien-1-yl)-2,2′,3,3′-tetramethoxy-1,1′-biphenyl. Molbank. 2025; 2025(2):M2018. https://doi.org/10.3390/M2018
Chicago/Turabian StyleDettori, Maria Antonietta, Davide Fabbri, Roberto Dallocchio, and Paola Carta. 2025. "5,5′-Di((E)-buta-1,3-dien-1-yl)-2,2′,3,3′-tetramethoxy-1,1′-biphenyl" Molbank 2025, no. 2: M2018. https://doi.org/10.3390/M2018
APA StyleDettori, M. A., Fabbri, D., Dallocchio, R., & Carta, P. (2025). 5,5′-Di((E)-buta-1,3-dien-1-yl)-2,2′,3,3′-tetramethoxy-1,1′-biphenyl. Molbank, 2025(2), M2018. https://doi.org/10.3390/M2018