
Synthesis of Dimethyl (Z)-((3-oxoindolin-2-ylidene) (aryl)methyl)phosphonates Through Tandem Cadogan and Arbuzov Reactions

 , , , , , and
, , , , , and 
Abstract
1. Introduction
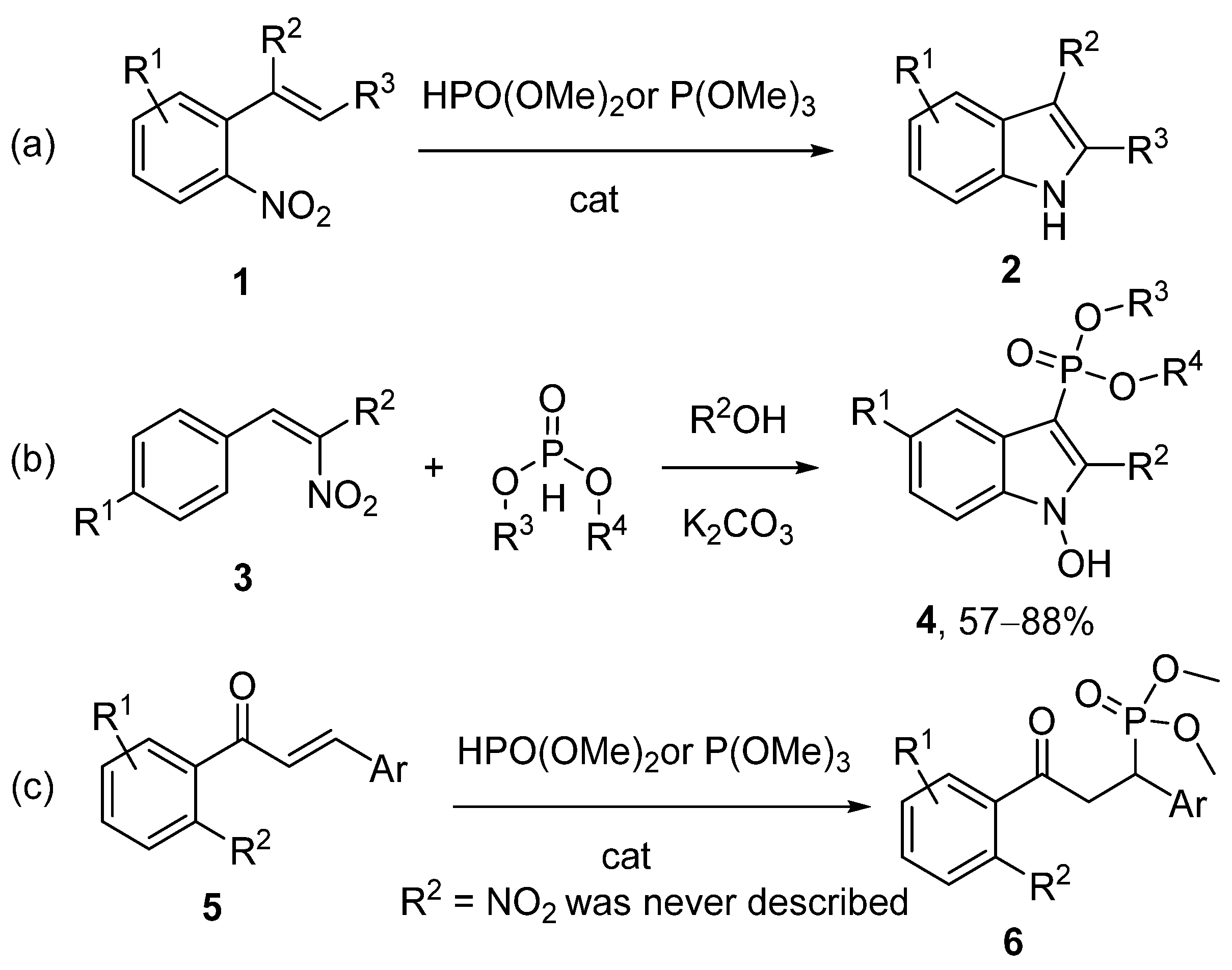
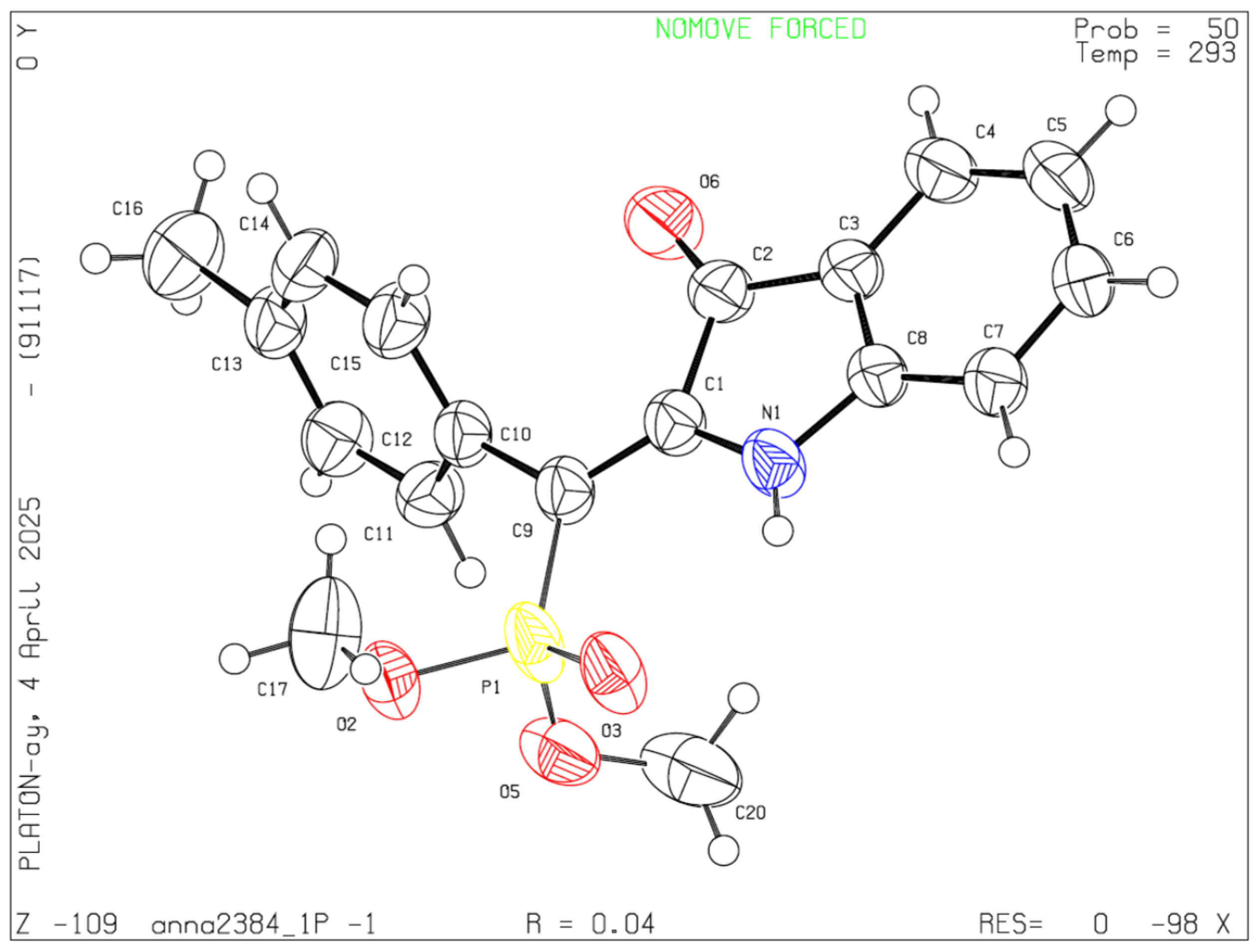
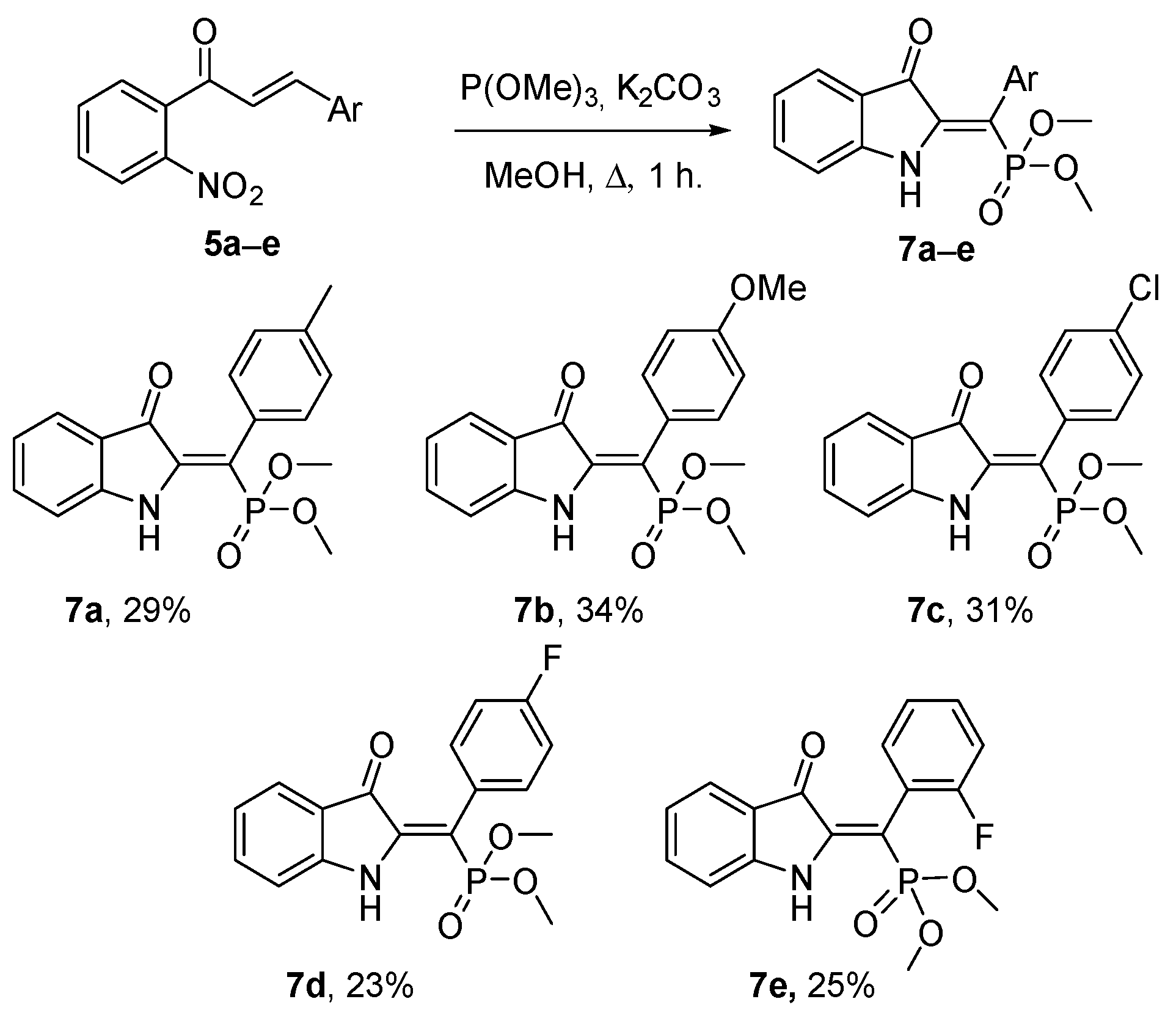
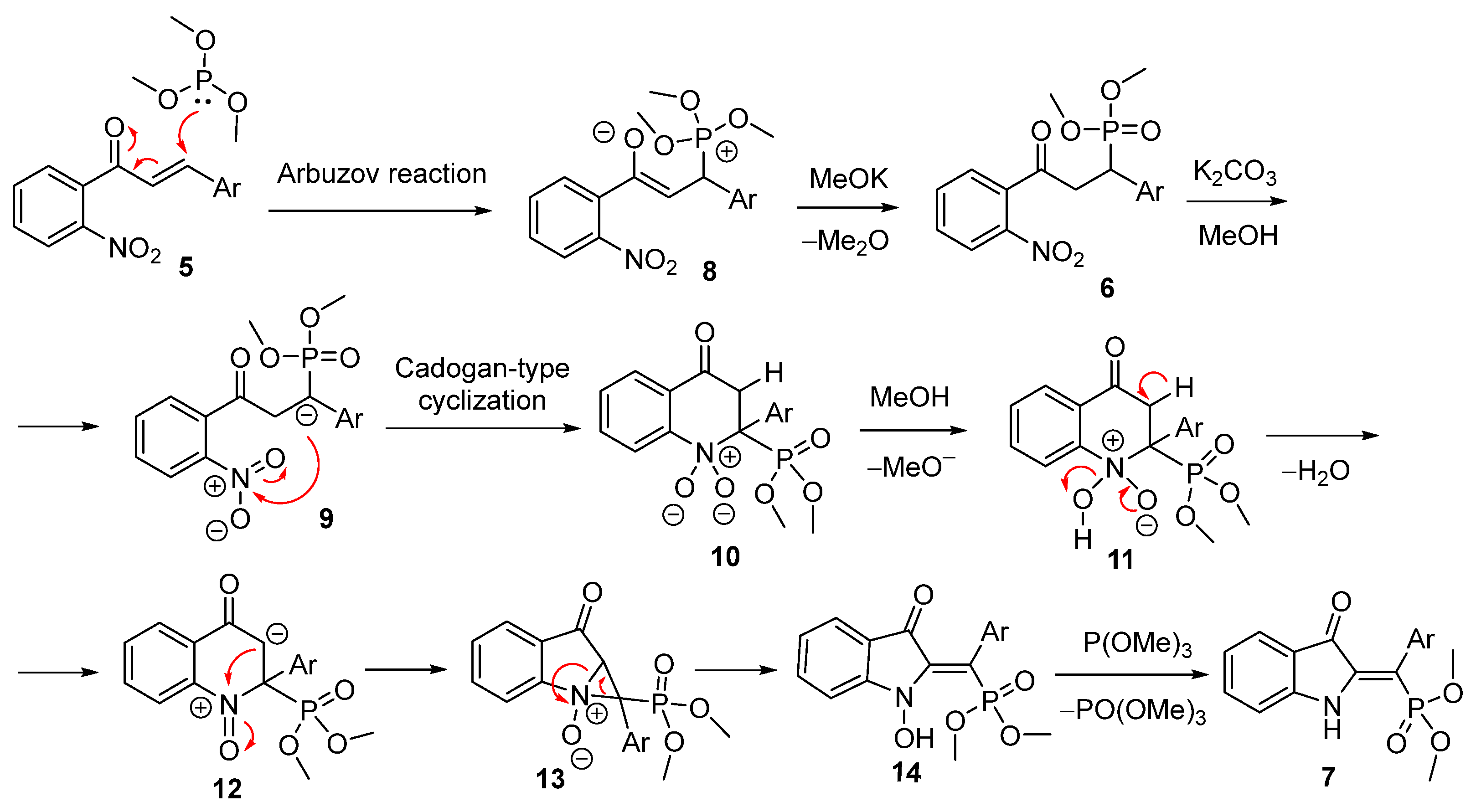
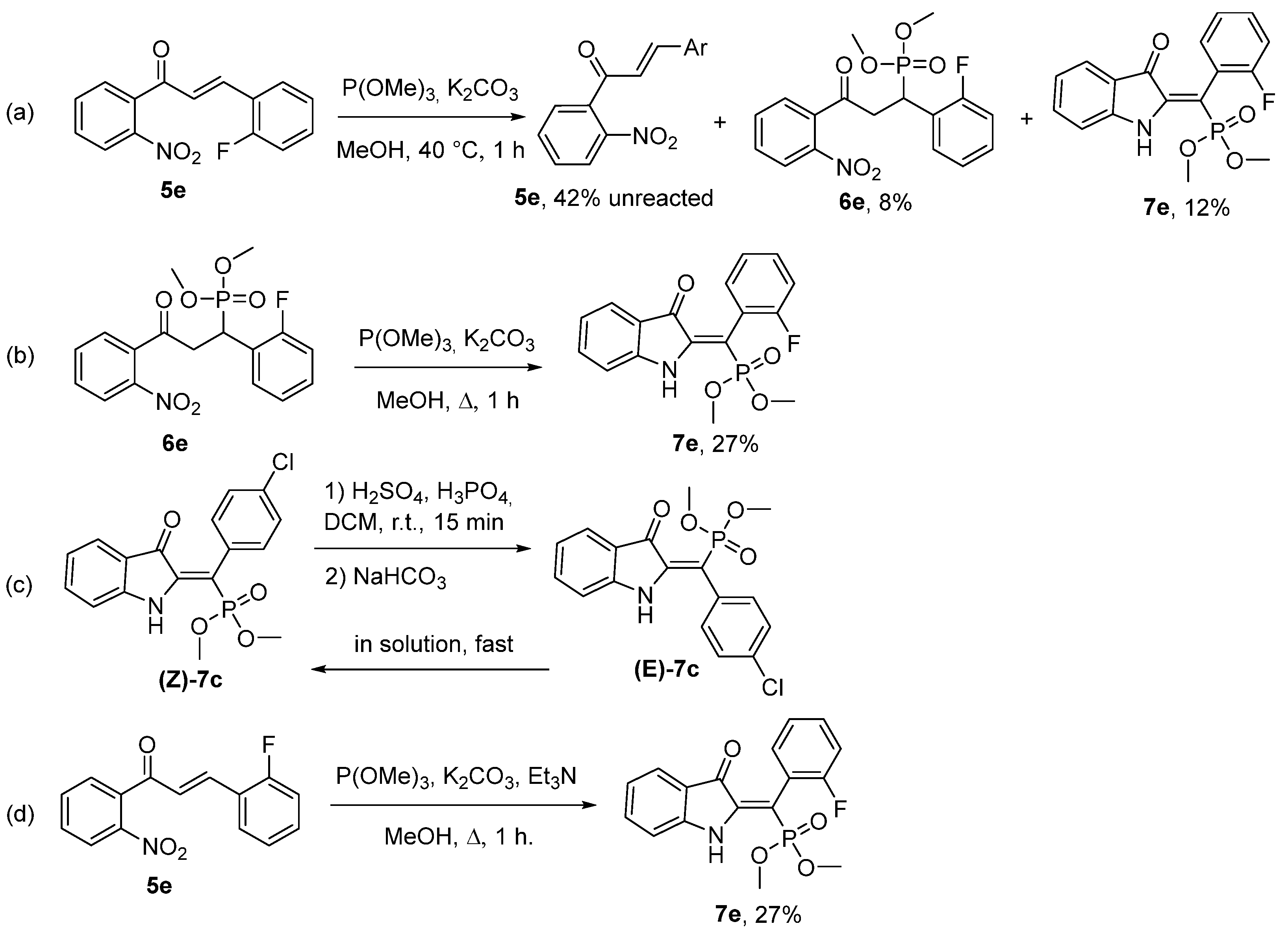
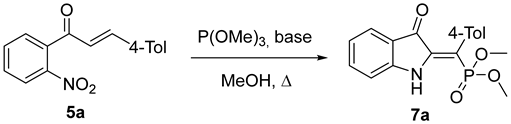
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Preparation of Dimethyl (Z)-((3-oxoindolin-2-ylidene)(aryl)methyl)phosphonates 7 (General Procedure)
3.3. Control Experiments: Preparation of Dimethyl (1-(2-fluorophenyl)-3-(2-nitrophenyl)-3-oxopropyl)phosphonate 6e (Scheme 4a)
3.4. Control Experiments: Preparation of Dimethyl (Z)-((2-fluorophenyl)(3-oxoindolin-2-ylidene)methyl)phosphonate 7e from Dimethyl (1-(2-fluorophenyl)-3-(2-nitrophenyl)-3-oxopropyl)phosphonate 6e (Scheme 4b)
3.5. Control Experiments: Z/E Isomerisation of Dimethyl (Z)-((4-chlorophenyl)(3-oxoindolin-2-ylidene)methyl)phosphonate (7c) in Acidic Medium (Scheme 4c)
3.6. Control Experiments: 2D TLC Observation of Z/E Isomerisation of Dimethyl (Z)-((4-chlorophenyl)(3-oxoindolin-2-ylidene)methyl)phosphonate (7c)
3.7. Control Experiments: Preparation of Dimethyl (Z)-((2-fluorophenyl)(3-oxoindolin-2-ylidene)methyl)phosphonate 7e in Presense of Et3N (Scheme 4d)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| DBU | 1,8-Diazabicyclo [5.4.0]undec-7-en |
| DCM | Dichloromethane |
| 2D TLC | Two-dimensional thin-layer chromatography |
References
- Omar, F.; Tareq, A.M.; Alqahtani, A.M.; Dhama, K.; Sayeed, M.A.; Emran, T.B.; Simal-Gandara, J. Plant-Based Indole Alkaloids: A Comprehensive Overview from a Pharmacological Perspective. Molecules 2021, 26, 2297. [Google Scholar] [CrossRef] [PubMed]
- Tudu, C.K.; Bandyopadhyay, A.; Kumar, M.; Radha; Das, T.; Nandy, S.; Ghorai, M.; Gopalakrishnan, A.V.; Proćków, J.; Dey, A. Unravelling the pharmacological properties of cryptolepine and its derivatives: A mini-review insight. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2023, 396, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Osafo, N.; Mensah, K.B.; Yeboah, O.K. Phytochemical and Pharmacological Review of Cryptolepis sanguinolenta (Lindl.). Schlechter. Adv. pharmacol. Pharm. Sci. 2017, 2017, 3026370. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, N.A.; Aksenov, A.V.; Kornienko, A.; De Carvalho, A.; Mathieu, V.; Aksenov, D.A.; Ovcharov, S.N.; Griaznov, G.D.; Rubin, M. A nitroalkane-based approach to one-pot three-component synthesis of isocryptolepine and its analogs with potent anti-cancer activities. RSC Adv. 2018, 8, 36980–36986. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J. Cadogan-Sundberg indole synthesis. In Name Reactions: A Collection of Detailed Mechanisms and Synthetic Applications, 5th ed.; Springer International Publishing: New York, NY, USA, 2014; pp. 102–103. [Google Scholar]
- Kaur, M.; Kumar, R. C-N and N-N bond formation via Reductive Cyclization: Progress in Cadogan /Cadogan-Sundberg Reaction. ChemistrySelect 2018, 3, 5330. [Google Scholar] [CrossRef]
- Reis, M.C.; Marín-Luna, M.; López, C.S.; Faza, O.N. Mechanism of the Molybdenum-Mediated Cadogan Reaction. ACS Omega 2018, 3, 7019–7026. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Liao, X.; Gao, Z. A Facile One-Pot Synthesis of 3-Dialkoxyphosphoryl- and 3-[Alkoxy(phenyl)phosphoryl]-1-hydroxyindoles. Synthesis 1990, 1990, 801–802. [Google Scholar] [CrossRef]
- Huang, Z.; Liu, W.; Li, S.; Yang, Y.; Guo, S.; Cai, H. Potassium Carbonate Promoted Nucleophilic Addition of Alkenes with Phosphites. Synlett 2020, 31, 1295–1297. [Google Scholar]
- Song, H.; Sun, Y.; Lu, C.; Zhao, B. Asymmetric Hydrophosphonylation of α,β-Unsaturated Ketones Catalyzed by Rare-Earth Metal Complexes Bearing Trost Ligands. J. Org. Chem. 2022, 87, 7747–7762. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Yang, J.; Yan, K.; Zhuang, X.; Yu, J.; Song, X.; Zhang, F.; Li, B.; Wen, J. Hydrophosphorylation of electron-deficient alkenes and alkynes mediated by convergent paired electrolysis. Chem. Comm. 2022, 58, 8238–8241. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Shao, N.; Li, F.-Z.; Yang, X.-C.; Wang, M.-C. Azetidine-derived dinuclear zinc catalyzed asymmetric phospha-Michael addition of dialkyl phosphite to α,β-unsaturated carbonyl compounds. Org. Biomol. Chem. 2017, 15, 9465–9474. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, N.A.; Aksenov, A.V.; Kurenkov, I.A.; Kirillov, N.K.; Aksenov, D.A.; Arutiunov, N.A.; Aksenova, D.S.; Rubin, M. One-Pot Synthesis of (E)-2-(3-Oxoindolin-2-ylidene)-2-arylacetonitriles. Molecules 2022, 27, 2808. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, N.A.; Aksenov, D.A.; Arutiunov, N.A.; Aksenova, D.S.; Aksenov, A.V.; Rubin, M. Unexpected cyclization of ortho-nitrochalcones into 2-alkylideneindolin-3-ones. RSC Adv. 2020, 10, 18440–18450. [Google Scholar] [CrossRef] [PubMed]
- Trawczyński, A.; Bujok, R.; Wróbel, Z.; Wojciechowski, K. Simple synthesis of pyrrolo[3,2-e]indole-1-carbonitriles. Beilstein J. Org. Chem. 2013, 9, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Hu, Z.; Zhang, X.; Dong, J.; Liu, J.-B.; Chen, D.-Z.; Xu, X. Tandem Synthesis of Pyrrolo[2,3-b]quinolones via Cadogen-Type Reaction. Org. Lett. 2017, 19, 5284–5287. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, N.A.; Aksenov, D.A.; Ganusenko, D.D.; Kurenkov, I.A.; Aksenov, A.V. A Diastereoselective Assembly of Tetralone Derivatives via a Tandem Michael Reaction and ipso-Substitution of the Nitro Group. J. Org. Chem. 2023, 88, 5639–5651. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||
|---|---|---|
| Entry | Base, 2 Equiv. | Yield, % |
| 1 | KOH | 0 |
| 2 | DBU | 0 |
| 3 | Et3N | 0 |
| 4 | MeONa | 14 |
| 5 | K2CO3 | 31 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aksenov, N.A.; Aksenov, D.A.; Ganusenko, D.D.; Kurlikov, A.E.; Barbolin, A.P.; Karaseva, P.S.; Aksenov, A.V. Synthesis of Dimethyl (Z)-((3-oxoindolin-2-ylidene) (aryl)methyl)phosphonates Through Tandem Cadogan and Arbuzov Reactions. Molbank 2025, 2025, M2002. https://doi.org/10.3390/M2002
Aksenov NA, Aksenov DA, Ganusenko DD, Kurlikov AE, Barbolin AP, Karaseva PS, Aksenov AV. Synthesis of Dimethyl (Z)-((3-oxoindolin-2-ylidene) (aryl)methyl)phosphonates Through Tandem Cadogan and Arbuzov Reactions. Molbank. 2025; 2025(2):M2002. https://doi.org/10.3390/M2002
Chicago/Turabian StyleAksenov, Nicolai A., Dmitrii A. Aksenov, Daniil D. Ganusenko, Alexander E. Kurlikov, Alexander P. Barbolin, Polina S. Karaseva, and Alexander V. Aksenov. 2025. "Synthesis of Dimethyl (Z)-((3-oxoindolin-2-ylidene) (aryl)methyl)phosphonates Through Tandem Cadogan and Arbuzov Reactions" Molbank 2025, no. 2: M2002. https://doi.org/10.3390/M2002
APA StyleAksenov, N. A., Aksenov, D. A., Ganusenko, D. D., Kurlikov, A. E., Barbolin, A. P., Karaseva, P. S., & Aksenov, A. V. (2025). Synthesis of Dimethyl (Z)-((3-oxoindolin-2-ylidene) (aryl)methyl)phosphonates Through Tandem Cadogan and Arbuzov Reactions. Molbank, 2025(2), M2002. https://doi.org/10.3390/M2002