
Syntheses of Chiral 2-Oxazolines from Isosorbide Epoxide Derivative


Abstract

{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
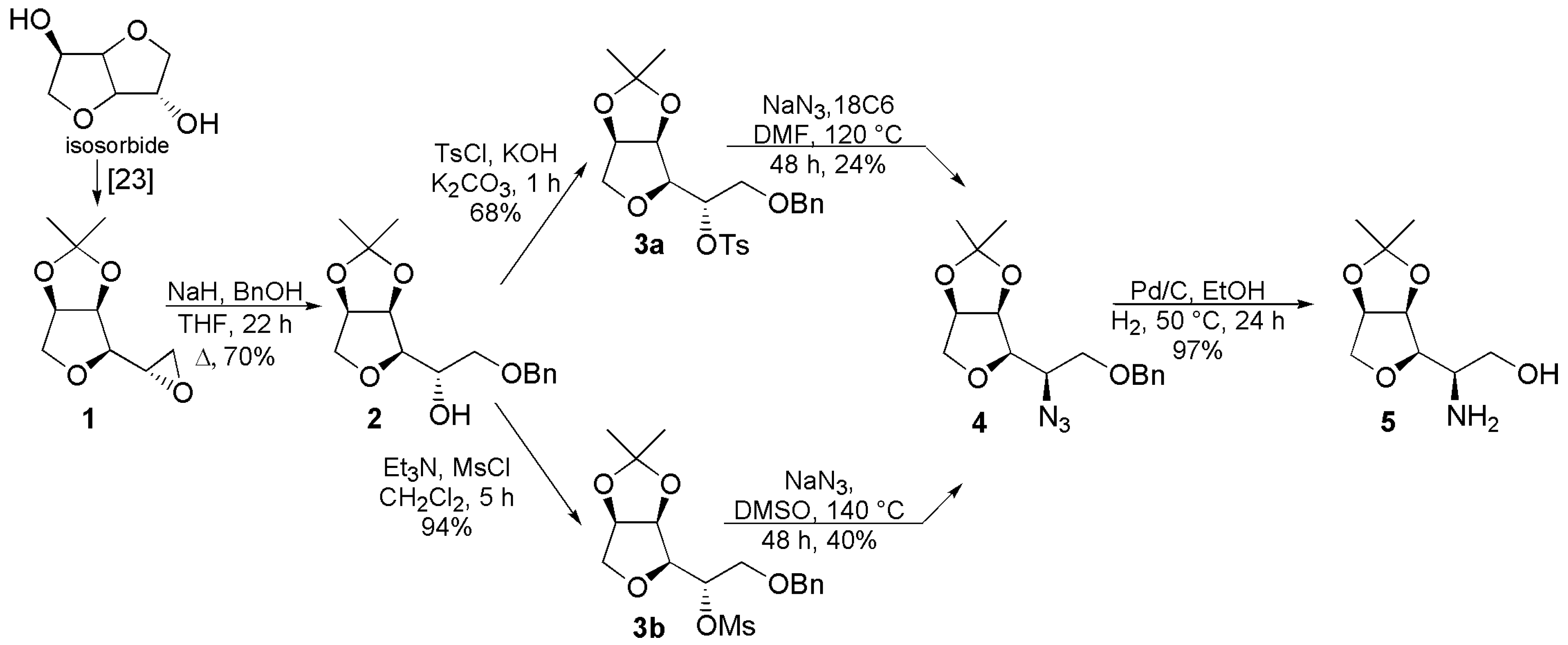
2.1. Formation of the Aminoalcohol
2.2. Synthesis of Oxazolines
3. Materials and Methods
3.1. General
3.2. Synthesis and Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andreasch, R. Zur Kenntniss des Allylhrnstoffs. Monatsh. Chem. 1884, 5, 33–46. [Google Scholar] [CrossRef]
- Meyers, A.I.; Temple, D.L. Syntheses via 2-oxazolines. II. Versatile synthesis of aliphatic carboxylic acids and esters. Mono-and dialkylation of acids masked by a simple protecting group. J. Am. Chem. Soc. 1970, 92, 6644–6646. [Google Scholar] [CrossRef]
- Meyers, A.I.; Temple, D.L.; Nolen, R.L.; Mihelich, E.D. Oxazolines. IX. Synthesis of homologated acetic acids and esters. J. Org. Chem. 1974, 39, 2778–2783. [Google Scholar] [CrossRef]
- Snieckus, V. Directed ortho metalation. Tertiary amide and O-carbamate directors in synthetic strategies for polysubstituted aromatics. Chem. Rev. 1990, 90, 879–933. [Google Scholar] [CrossRef]
- Mahand, S.N.; Aliakbarzadeh, S.; Moghaddam, A.; Moghaddam, A.S.; Kruppke, B.; Nasrollahzadeh, M.; Khonakdar, H.A. Polyoxazoline: A review article from polymerization to smart behaviors and biomedical applications. Eur. Polym. J. 2022, 178, 111484. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Lizos, D.E.; Kim, D.W.; Schlawe, D.; de Noronha, R.G.; Longbottom, D.A.; Rodriquez, M.; Bucci, M.; Cirino, G. Total synthesis and biological evaluation of halipeptins A and D and analogues. J. Am. Chem. Soc. 2006, 128, 4460–4470. [Google Scholar] [CrossRef]
- Pirrung, M.C.; Tumey, L.N.; McClerren, A.L.; Raetz, C.R.H. High-throughput catch-and-release synthesis of oxazoline hydroxamates. Structure-activity relationships in novel inhibitors of Escherichia coli LpxC: In vitro enzyme inhibition and antibacterial properties. J. Am. Chem. Soc. 2003, 125, 1575–1586. [Google Scholar] [CrossRef]
- Bode, H.B.; Irschik, H.; Wenzel, S.C.; Reichenbach, H.; Müller; Höfle, G. The Leupyrrins: A Structurally Unique Family of Secondary Metabolites from the Myxobacterium Sorangium cellulosum. J. Nat. Prod. 2003, 66, 1203–1206. [Google Scholar] [CrossRef]
- Kline, T.; Andersen, N.H.; Harwood, E.A.; Bowman, J.; Malanda, A.; Endsley, S.; Erwin, A.L.; Doyle, M.; Fong, S.; Harris, A.L.; et al. Potent, novel in vitro inhibitors of the Pseudomonas aeruginosa deacetylase LpxC. J. Med. Chem. 2002, 45, 3112–3129. [Google Scholar] [CrossRef]
- Bergeron, R.J.; Xin, M.G.; Weimar, W.R.; Smith, R.E.; Wiegand, J. Significance of asymmetric sites in choosing siderophores as deferration agents. J. Med. Chem. 2001, 44, 2469–2478. [Google Scholar] [CrossRef]
- Marques de Oliveira, A.R.; Simonelli, F.; de Assis Marques, F.; Clososki, G.C.; Oliveira, A.M.; Lenz, C.A. 2-oxazolinas quiras: Algumas plicaçoes como indutores de assimetria em reaçoes organicas. Quim. Nova 1999, 22, 854–862. [Google Scholar] [CrossRef]
- Meyers, A.I. Chiral oxazolines and their legacy in asymmetric carbon-carbon bond-forming reactions. J. Org. Chem. 2005, 70, 6137–6151. [Google Scholar] [CrossRef]
- McManus, H.A.; Guiry, P.J. Recent Developments in the Application of Oxazoline-Containing Ligands in Asymmetric Catalysis. Chem. Rev. 2004, 104, 4151–4202. [Google Scholar] [CrossRef]
- Hargaden, G.C.; Guiry, P.J. Recent Applications of Oxazoline-Containing Ligands in Asymmetric Catalysis. Chem. Rev. 2009, 109, 2505–2550. [Google Scholar] [CrossRef]
- Connon, R.; Roche, B.; Rokade, B.V.; Guiry, P.J. Further developments and applications of oxazoline-containing ligands in asymmetric catalysis. Chem. Rev. 2021, 121, 6373–6521. [Google Scholar] [CrossRef]
- For example Saiyed, A.S.; Bedekar, A.V. Amino oxazolines as a new class of organocatalyst for the direct intermolecular asymmetric aldol reaction between acetone and aromatic aldehydes. Tetrahedron Asymmetry 2013, 24, 1035–1041. [Google Scholar] [CrossRef]
- van Es, D.S. Rigid Biobased Building Blocks: Current Developments and Outlook. J. Renew. Mater. 2013, 1, 61–72. [Google Scholar] [CrossRef]
- Frump, J.A. Oxazolines. Their Preparation, Reactions, and Applications. Chem. Rev. 1971, 71, 483–504. [Google Scholar] [CrossRef]
- Gant, T.G.; Meyers, A.I. The chemistry of 2-oxazolines (1985–present). Tetrahedron 1994, 50, 2297–2360. [Google Scholar] [CrossRef]
- Hansen, J.F.; Cooper, C.S. Preparation and alkylation of a new chiral oxazoline from L-serine. J. Org. Chem. 1976, 41, 3219–3220. [Google Scholar] [CrossRef]
- Meyers, A.I.; Whitten, C.E. Oxazolines XXIV: Chiral Oxazolines and Thiazolines from L-Serine and L-Cysteine. Their Potential Use in Asymmetric Synthesis. Heterocycles 1976, 4, 1687–1692. [Google Scholar] [CrossRef]
- Brunner, H.; Berghofer, J. Enantioselektive Katalyse 97. Optisch aktive Salicyloxazolin-Liganden in der enantioselektiven Kupfer-katalysierten Cyclopropanierung. J. Organomet. Chem. 1995, 501, 161–166. [Google Scholar] [CrossRef]
- Boiaryna, L.; Guillarme, S.; Saluzzo, C. 5(S)-((3aR,4R,6aR)-2,2-Dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-2-phenyl-4,5-dihydrooxazole. Molbank 2024, 2024, M1843. [Google Scholar] [CrossRef]
- Kazemi, F.; Massahb, A.R.; Javaherianc, M. Chemoselective and scalable preparation of alkyl tosylates under solvent-free conditions. Tetrahedron 2007, 63, 5083–5087. [Google Scholar] [CrossRef]
- Black, D.S.C.; Wade, M.J. Synthetic studies related to mycobactins. I. Synthesis of 2-(2′-hydroxyphenyl)-2-oxazoline-4-carboxylic acid and related compounds. Aust. J. Chem. 1972, 25, 1797–1810. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kadraoui, M.; Guillarme, S.; Saluzzo, C. Syntheses of Chiral 2-Oxazolines from Isosorbide Epoxide Derivative. Molbank 2025, 2025, M1966. https://doi.org/10.3390/M1966
Kadraoui M, Guillarme S, Saluzzo C. Syntheses of Chiral 2-Oxazolines from Isosorbide Epoxide Derivative. Molbank. 2025; 2025(1):M1966. https://doi.org/10.3390/M1966
Chicago/Turabian StyleKadraoui, Mohammed, Stéphane Guillarme, and Christine Saluzzo. 2025. "Syntheses of Chiral 2-Oxazolines from Isosorbide Epoxide Derivative" Molbank 2025, no. 1: M1966. https://doi.org/10.3390/M1966
APA StyleKadraoui, M., Guillarme, S., & Saluzzo, C. (2025). Syntheses of Chiral 2-Oxazolines from Isosorbide Epoxide Derivative. Molbank, 2025(1), M1966. https://doi.org/10.3390/M1966