1. Introduction
Anions play an instrumental role in a wide range of biological and environmental processes, and, as a result, their recognition by synthetic anion receptors has been an active area of research since the late 1960s [
1]. The recognition of phosphates is of particular interest due to their high biological relevance, being involved in processes such as energy storage and signal transduction [
2,
3] and in diseases such as hyperphosphatemia [
4]. Furthermore, their overabundance in environmental systems has potentially adverse outcomes [
5].
Synthetic anion receptors can use different types of interactions, such as hydrogen bonds, halogen bonds, and anion–π interactions. The most effective anion receptors are obtained by the combined action of multiple pre-organized interacting groups [
6,
7,
8]. The development of such receptors requires rigid molecular scaffolds, preferably functionalized with amines that can be turned into binding groups such as amides, ureas, thioureas, or squaramides. The distance between these amine groups and their orientation determines the size of the binding site of the receptor.
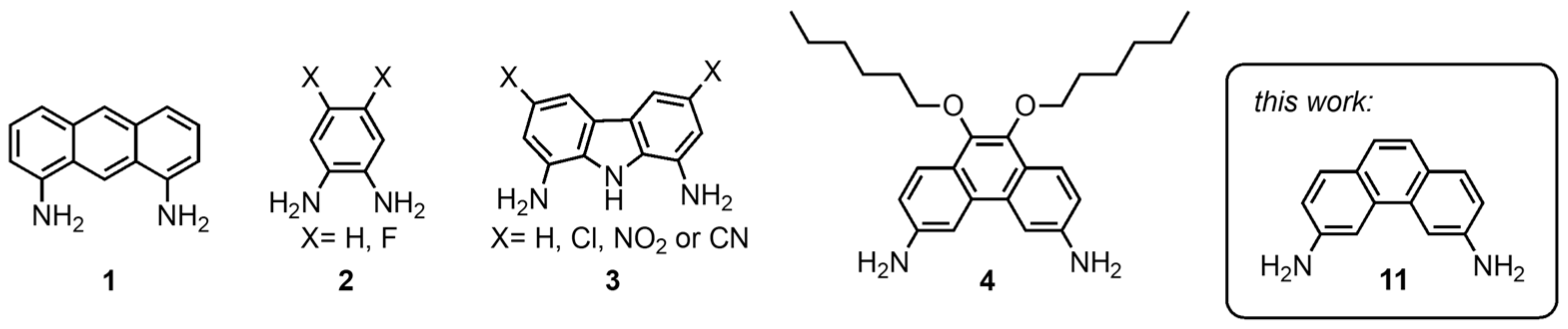
For smaller anions such as chloride, a wealth of molecular scaffolds has been employed [
9,
10,
11], including 1,8-diaminoanthrancene (
1) [
12] and
ortho-phenylenediamines (
2) [
13] (
Figure 1). For larger anions with a more complex geometry such as phosphate, fewer simple scaffolds are available [
14]. An example is the diaminocarbazoles reported by Chmielewski et al. (
3) [
15,
16]. In our previous work on macrocyclic anion receptors, we have described the synthesis of a 3,6-diaminophenanthrene featuring two hexyloxy-substituents (
4) for the synthesis of macrocyclic receptors for phosphate and pyrophosphate [
17]. Compound
4 was obtained via a Buchwald–Hartwig coupling and its synthesis route featured several difficult and tedious purification steps. Additionally, the two hexyloxy-substituents may not be necessary nor desired in many cases.
In the present work, we present a facile synthetic route to the basic 3,6-diaminophenanthrene building block (11) without additional substituents, using readily available starting materials and reagents. The use of this basic building block for the synthesis of a bis-urea anion receptor is also demonstrated.
2. Results and Discussion
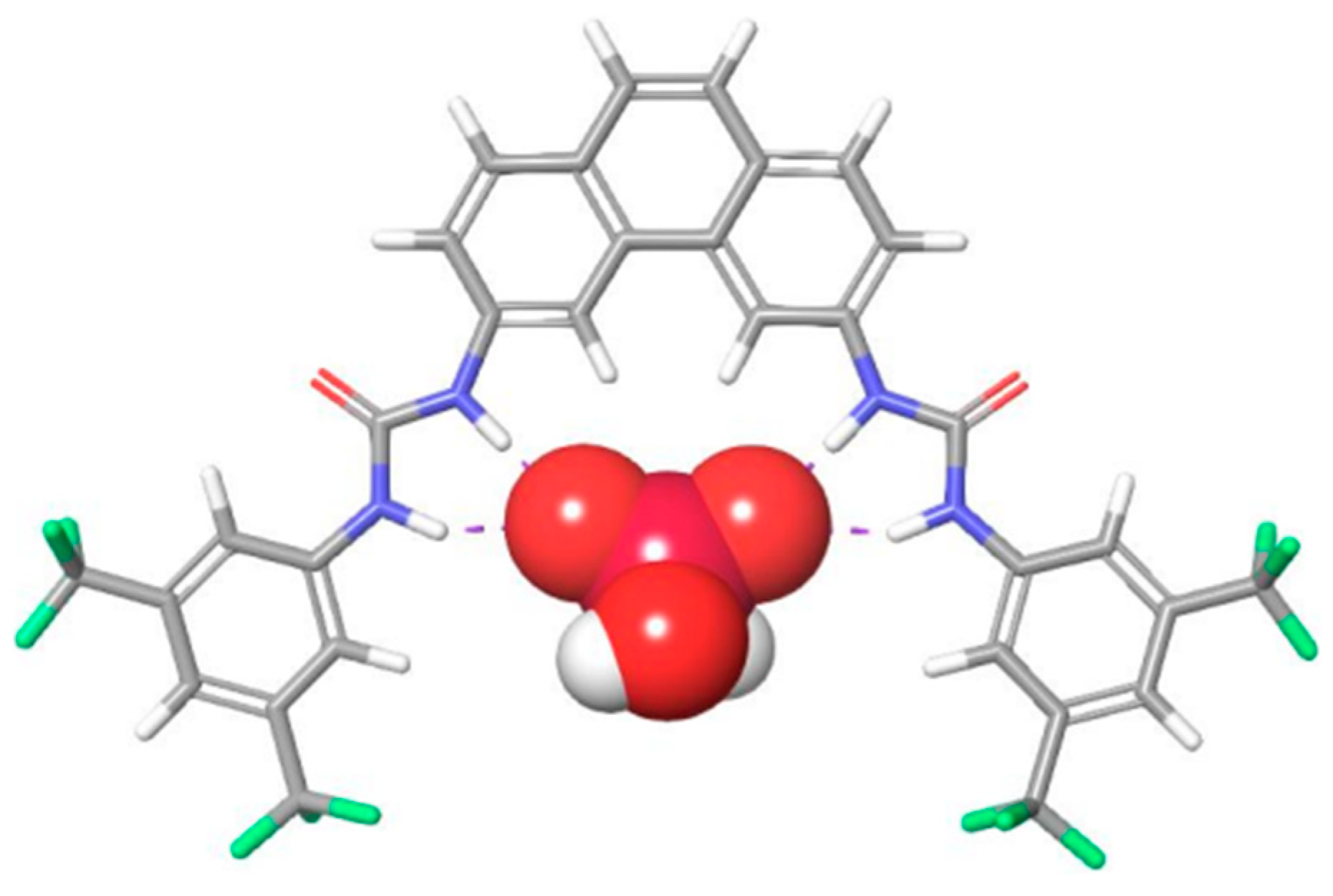
As part of a larger project aimed at the synthesis of receptors capable of binding phosphate and phosphate derivatives, we set out to develop a molecular scaffold well-suited for the binding of such phosphates. Molecular mechanics modeling (
Figure 2) suggested that a phenanthrene appended with binding moieties at the 3- and 6-positions would provide us with receptors that would have a binding site with a size suitable to accommodate phosphate, as well as a rigid backbone to help ensure a good orientation of the binding moieties and avoid intramolecular H-bonding between the two urea groups.
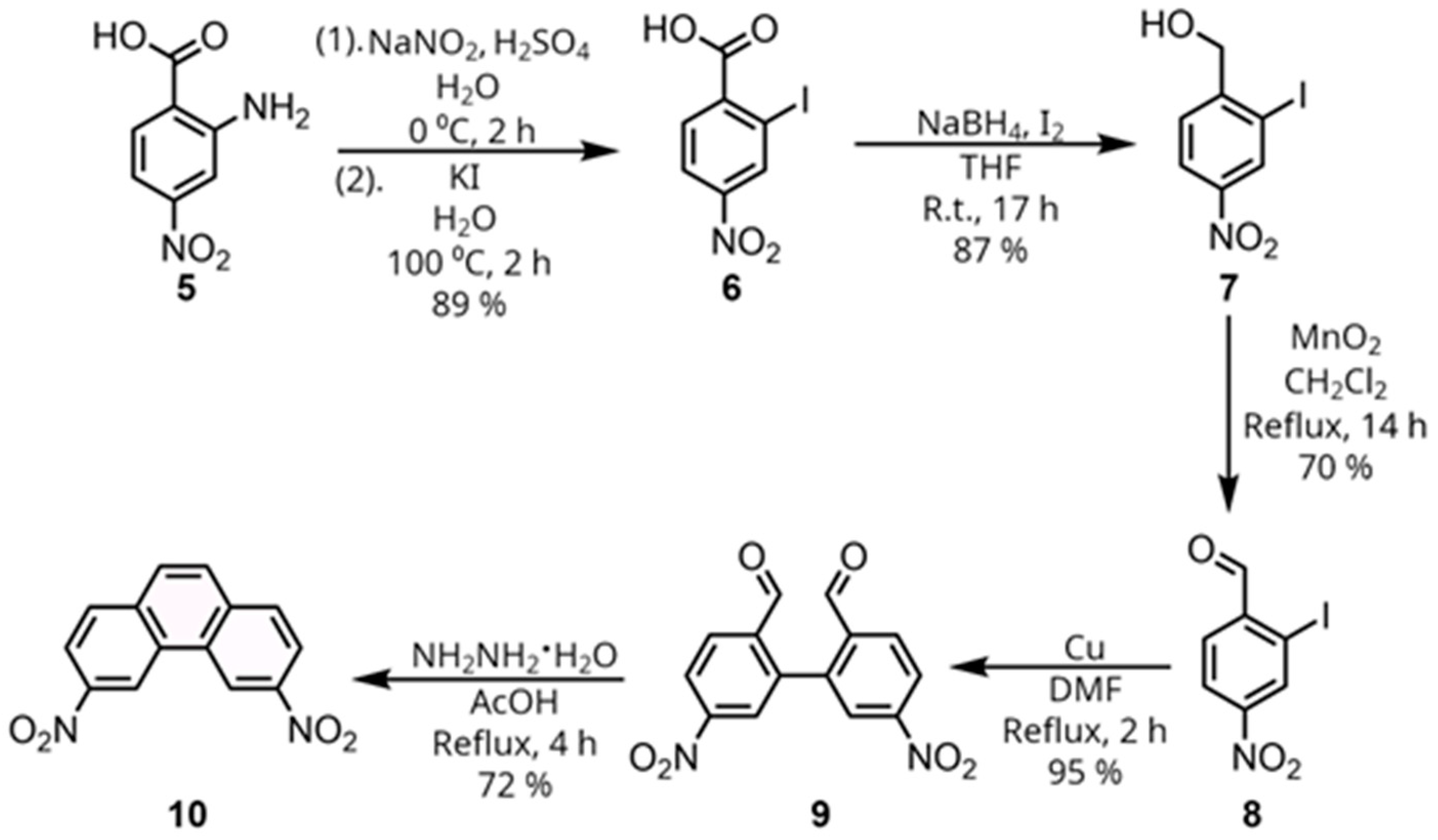
The molecular scaffold required to obtain such a receptor is the corresponding 3,6-diaminophenanthrene (
11), which was reported in 1968 by Staab et. al. [
18], but has seen little utilization since. A synthesis of the precursor 3,6-dinitrophenanthrene (
10) from 2-iodo-4-nitrobenzaldehyde (
8) was reported in 1958 by Bacon and Lindsay [
19], as outlined in
Scheme 1, and was chosen for our route to diamine
11. To obtain 2-iodo-4-nitrobenzaldehyde (
8), we started from commercially available 4-nitroanthranillic acid (
5). The first step is a Sandmeyer reaction, starting with a diazotization, followed by a reaction with potassium iodide in a one-pot procedure to yield the corresponding 2-iodo-4-nitrobenzoic acid (
6) [
20]. Next, this carboxylic acid was reduced using diborane generated from NaBH
4 and I
2 to obtain (2-iodo-4-nitrophenyl)methanol (
7) [
21]. The alcohol could then be oxidized using manganese dioxide to obtain 2-iodo-4-nitrobenzaldehyde (
8) [
21]. A classical Ullmann coupling of 2-iodo-4-nitrobenzaldehyde (
8) using copper in refluxing DMF then afforded the corresponding biphenyl dicarbaldehyde (
9). Finally, a reductive cyclization using hydrazine hydrate in refluxing glacial acetic acid was employed to form the phenanthrene ring system and obtain the desired 3,6-dinitrophenanthrene (
10). Notably, only the reduction from the carboxylic acid (
6) to the alcohol (
7) required a chromatographic separation, with the other steps only requiring either filtration or recrystallization.
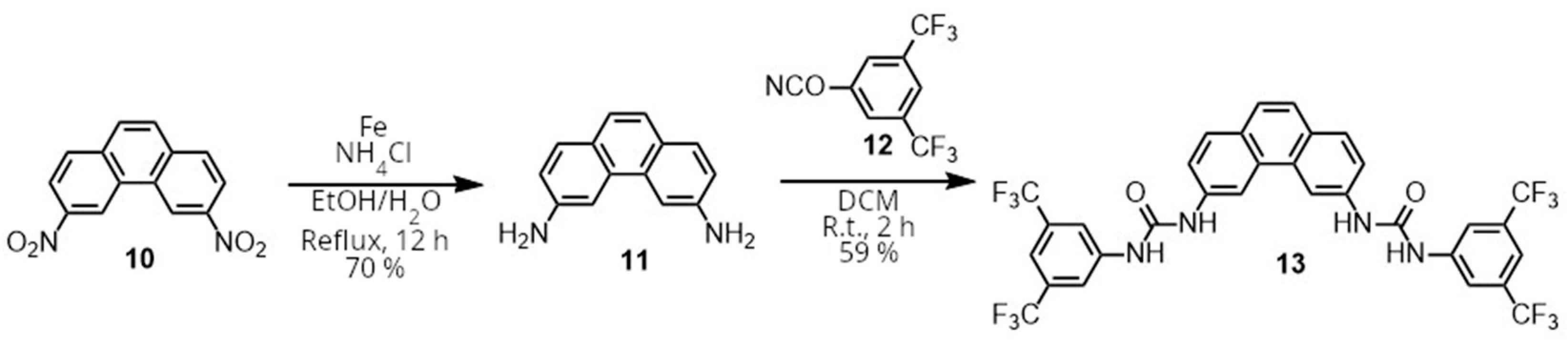
Having obtained 3,6-dinitrophenanthrene (
10), attention was now turned to the reduction of the nitro groups (
Scheme 2). Due to the expected poor stability of the phenanthrene diamine (
11), mild methods of reduction such as NH
2NH
2•H
2O and Pd/C or NaBH
4 and NiCl
2 were tested. However, while these reagents led to a reduction, very significant amounts of decomposition products were observed, even with reaction times as low as 30 min. Eventually, we found that using iron powder and NH
4Cl in a refluxing mixture of ethanol and water afforded the diamine (
11) with minimal decomposition and an easy, fast workup in 70% yield. Upon obtaining the pure amine, its very poor stability in the absence of additional stabilizing agents was confirmed, as the initially light brown solid would turn completely black in a few hours, even when stored under a vacuum in the absence of light. Acetonitrile solutions of
11 used for NMR analysis would also change from very slightly colored transparent solutions to completely black and opaque solutions in a few hours.
Therefore, the diamine was used directly after its synthesis to form receptor 13 by reacting it with isocyanate 12. The desired compound precipitated from the reaction mixture, requiring only filtration and washing to obtain the pure compound 13.
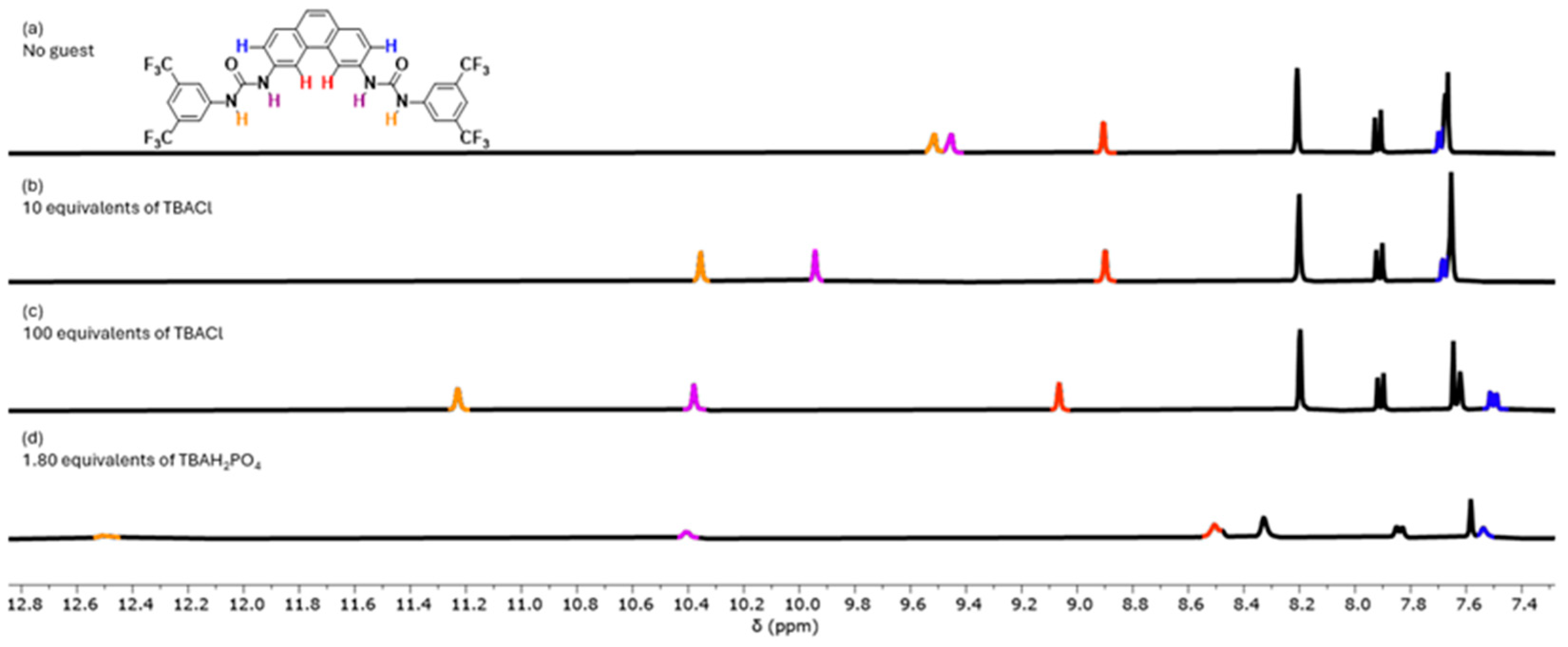
The receptor was screened for anion binding with tetrabutylammonium (TBA) salts of Cl
− and H
2PO
4− in DMSO-d
6 with 0.5% H
2O. Clear downfield changes in the chemical shifts of the signals corresponding to urea H-atoms in the binding site were observed, indicative that both anions interact with
13 (
Figure 3). The full titrations are presented in the supporting information (
Figures S20 and S21) and show clear indications that the stoichiometry is more complex than a 1:1 receptor-to-anion interaction. For the titrations with Cl
−, the changes in chemical shifts could be fitted to a 1:2 binding model (
Figure S20), which gave affinity constants K
a(1:1) = 87 M
−1 and a K
a(1:2) = 17 M
−1. This K
a(1:1) value is similar to the one obtained for a simple urea with bis(CF
3)phenyl groups [
22], which is not surprising, as Cl
− is too small to interact with both urea groups at the same time (
Figures S22 and S23). For H
2PO
4−, the broadening of both NH signals over the course of the titration and the complex behavior of other signals prevented quantification of the interaction of
13 with H
2PO
4− (
Figure S21). However, the sharpening of the signals with around 1.8 equivalents of H
2PO
4− could indicate that
13 can bind two H
2PO
4− anions. Molecular modeling confirmed that a H
2PO
4− dimer [
23] would fit in the binding site of
13 (
Figure S24).
3. Materials and Methods
All chemicals used were bought through either Sigma Aldrich, VWR, or Fluorochem and were used as received unless otherwise stated. Dry solvents were either bought as anhydrous solvents through commercial suppliers or dried over molecular sieves for at least 48 h before use. Column chromatography was carried out using silica gel with a 60 Å pore size and a particle size of 40–63 µm for flash column chromatography (FCC). TLC analysis was carried out on aluminum-backed silica gel plates visualized by a UV lamp or, when appropriate, permanganate or ninhydrin stains. Melting points were measured on a Cole-Parmer Stuart SMP-200. IR spectroscopy was carried out on a Shimadzu IRSpirit-X FTIR spectrometer. NMR analysis was carried out on either a 400 MHz Varian spectrometer, a 400 MHz Jeol spectrometer, or a 600 MHz Jeol spectrometer. Chemical shifts are reported in ppm relative to the residual solvent signal of the deuterated solvent and the coupling constants are reported in Hz. ESI-MS Mass spectrometry was carried out on a Thermo Finnigan LCQ-DecaXP and HRMS was carried out on an Agilent QTOF 6520 by electron spray ionization.
3.1. Synthesis of 2-Iodo-4-nitrobenzoic Acid (6)
Following previously reported large-scale synthesis of this compound [
20]. To a stirred solution of 2-amino-4-nitrobenzoic acid (5.0 g, 27.45 mmol) in water (80 mL) cooled on an ice bath was added concentrated sulfuric acid (5.55 mL) and a solution of sodium nitrite (2.0 g, 29.65 mmol) in water (14 mL), added dropwise while keeping the temperature less than 5 °C. The mixture was stirred at 0–5 °C for 2 h, and then a solution of potassium iodide (6.290 g, 37.88 mmol in water (14 mL) was added over 10 min maintaining the temperature below 5 °C. The mixture was heated to 100 °C for 2 h and then cooled in an ice bath. The formed precipitate was collected by filtration, taken up in ether (55 mL), and washed with 20% aqueous thiosulfate solution (3 × 20 mL). The organic layer was dried with MgSO
4 and evaporated to dryness under reduced pressure. The solid residue was recrystallized from water to give 2-iodo-4-nitrobenzoic acid (7.0 g, 89%) as an orange solid. Mp: 143–145 °C (lit. 142–143 °C [
20]).
1H NMR (400 MHz, chloroform-d) δ 8.86 (d,
J = 2.2, 1H), 8.29 (dd,
J = 8.6, 2.2 1H), 8.11 (d,
J = 8.6, 1H).
13C NMR (101 MHz, chloroform-d) δ 168.78, 149.48, 139.07, 136.56, 132.31, 122.97, 94.27. The analytical data were found to match those of the literature.
3.2. Synthesis of (2-Iodo-4-nitrophenyl)methanol (7)
Following the general procedure reported in [
21]. A flame-dried round bottom flask under an argon atmosphere was charged with 2-iodo-3-methylbenzoic acid (1 g, 3.41 mmol), THF (22.0 mL), and NaBH
4 (0.516 g, 13.65 mmol) and the resulting solution cooled to 0 °C. A solution of I
2 (0.866 g, 3.41 mmol) in THF (11.0 mL) was added dropwise to the reaction mixture at 0 °C and the reaction mixture was stirred at room temperature for 17 h. Aqueous HCl (1 M, 72.5 mL) was added to the reaction mixture and the aqueous phase was extracted with Et
2O three times. The combined organic layer was washed with brine, dried over Na
2SO
4, filtrated, and concentrated. The obtained residue was purified by FCC (1:1 ethyl acetate/heptane) to yield the desired compound as an orange solid (828 mg, 87%). Mp: 94–96 °C (lit. 96.4–97.5 °C [
21]).
1H NMR (400 MHz, chloroform-d) δ 8.65 (d,
J = 2.3, 1H), 8.25 (dd,
J = 8.6, 2.3, 1H), 7.70 (d,
J = 8.83 1H), 4.73 (s, 2H).
13C NMR (101 MHz, chloroform-d) δ 149.90, 146.91, 133.93, 127.89, 123.45, 95.23, 68.80. The analytical data were found to match that of the literature.
3.3. Synthesis of 2-Iodo-4-nitrobenzaldehyde (8)
Following the general procedure reported in [
21]. A mixture of (2-iodo-4-nitrophenyl)methanol
7 (200 mg, 716 µmol) and MnO
2 (1.93 g, 22.15 mmol) in CH
2Cl
2 (10.0 mL) was refluxed for 14 h. After cooling to rt, the mixture was filtered over a celite pad, the pad washed with additional CH
2Cl
2 (3 × 10.0 mL), and the solvent removed under reduced pressure to give the desired aldehyde (140 mg, 70%). Mp: 82–85 °C (lit. 84.5 °C [
19]).
1H NMR (400 MHz, chloroform-d) δ 10.14 (d,
J = 0.78, 1H), 8.79 (d,
J = 2.1, 1H), 8.3 (ddd,
J = 8.36, 2.2, 0.78 1H), 8.01 (d,
J = 8.5, 1H).
13C NMR (101 MHz, chloroform-d) δ 193.88, 146.36, 139.10, 135.56, 130.86, 123.60, 99.14. The analytical data were found to match that of the literature.
3.4. Synthesis of 5,5′-Dinitro-[1,1′-biphenyl]-2,2′-dicarbaldehyde (9)
Based on the procedure published by Bacon and Lindsay [
19], 2-iodo-4-nitrobenzaldehyde
8 (130 mg, 469 µmol) was mixed together with copper powder (89 mg, 1.41 mmol) and DMF (10 mL) in a flame-dried three-necked round bottom flask fitted with a reflux condenser, a nitrogen balloon, and a vacuum outlet. Three vac/nitrogen cycles were performed, and the mixture heated to reflux for 2 h. After cooling to room temperature, the mixture was filtered through a pad of celite, and the pad washed with CH
2Cl
2 (2 × 15 mL). The combined organic phase was evaporated in vacuo to yield the biphenyl compound as a light-brown solid, which was used for the next step without further purification (66 mg, 95%). Mp: 201–203 °C (lit. 203.5 °C [
19]). FT-IR (cm
−1): 1349,1526, 1681, 1690, 2854.
1H NMR (400 MHz, DMSO-d
6) δ 9.86 (s, 2H), 8.51 (dd,
J = 8.4, 2.4, 2H), 8.36 (d,
J = 2.3, 2H), 8.28 (d,
J = 8.4, 2H).
13C NMR (101 MHz, DMSO-d
6) δ 191.55, 150.09, 140.08, 138.65, 131.87, 127.03, 124.53. HRMS(+) calc. for C
14H
9N
2O
6+: 301.0455 found: 301.0470.
3.5. Synthesis of 3,6-Dinitrophenanthrene (10)
Based on the procedure published by Bacon and Lindsay [
19], 5,5′-dinitro-[1,1′-biphenyl]-2,2′-dicarbaldehyde
9 (351 mg, 1.17 mmol) was dissolved in glacial acetic acid (3 mL) and heated to reflux. Hydrazine hydrate (116 µL, 2.34 mmol) in glacial acetic acid (1 mL) was added dropwise, and the mixture heated for a further 4 h. The mixture was cooled to room temperature and left in a fridge overnight to precipitate. The following morning, the formed precipitate was filtered and the mother liquor concentrated to roughly 1 mL and left in the fridge for one additional day to precipitate further. The resulting precipitate was filtered off, and the combined filtrides dried in vacuo to yield the desired compound as brown-colored needle-like crystals (226 mg, 72%). Mp: Decomposition above 260 °C (lit. 282–282.5 [
19]). FT-IR (cm
−1): 1340, 1502, 1508.
1H NMR (400 MHz, DMSO-d
6) δ 9.80 (d,
J = 2.2, 2H), 8.52 (dd,
J = 8.8, 2.2, 2H), 8.37 (d,
J = 8.8, 2H), 8.28 (s, 2H).
13C NMR (101 MHz, DMSO-d
6) δ 147.07, 136.22, 131.28, 130.59, 130.06, 122.34, 120.39. HRMS(+) calc. for C
14H
9N
2O
4+: 269.0557 found: 269.0560.
3.6. Synthesis of 3,6-Diaminophenanthrene (11)
3,6-dinitrophenanthrene 10 (60 mg, 223 µmol) was suspended in ethanol (3 mL) and water (0.8 mL). Iron powder (86 mg, 1.54 mmol) and ammonium chloride (60 mg, 1.12 mmol) were added and the mixture was kept at reflux for 12 h. The reaction was then filtered through a pad of celite and the pad washed with H2O/EtOH (20%, 5 mL) and ethyl acetate (5 mL). The filtrate was then diluted with additional ethyl acetate (10 mL) and brine (10 mL). The two phases were separated and the aqueous phase was extracted twice more with ethyl acetate (10 mL). The combined organic phase was washed with saturated Na2CO3, water, and brine and dried with MgSO4 to yield the crude diamine as a brown solid (33 mg, 71%). FT-IR (cm−1): 1621, 1655, 3232, 3337. 1H NMR (400 MHz, Acetonitrile-d3) δ 7.68 (d, J = 2.2, 2H), 7.59 (d, J = 8.4, 2H), 7.31 (s, 2H), 6.97 (dd, J = 8.5, 2.2, 2H), 4.43 (s, 4H). 13C NMR (101 MHz, Acetonitrile-d3) δ 147.39, 131.83, 130.38, 126.10, 123.53, 117.70, 105.92. ESI-MS (+) calc. for C14H13N2+: 209.11 found: 209.13.
3.7. 1,1′-(Phenanthrene-3,6-diyl)bis(3-(3,5-bis(trifluoromethyl)phenyl)urea) (13)
A flame-dried 5 mL round bottom flask fitted with a septum with an argon balloon was vented to the atmosphere to replace the air with argon. 3,6-diaminophenanthrene 11 (26 mg, 124 µmol) was transferred to the flask as a CH2Cl2 (4 mL) solution using dry CH2Cl2. Next, the 3,5- bis(trifluoromethyl)phenyl isocyanate (43 µL, 249 µmol) was added dropwise over 2 min, and the resulting mixture left to stir for two hours. The resulting precipitate was filtered and washed with cold CH2Cl2 (3 × 3 mL) and dried in vacuo to yield the expected bis-urea as a tan solid (53 mg, 59%) Mp: above 300 °C. FT-IR (cm−1): 1474, 1573, 1665, 3123, 3321. 1H NMR (400 MHz, DMSO-d6) δ 9.50 (s, 2H), 9.44 (s, 2H), 8.90 (d, J = 2.2, 2H), 8.21 (d, J = 1.6, 4H), 7.92 (d, J = 8.6, 2H), 7.70 (dd, J = 8.6, 2.1, 2H), 7.68–7.66 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 152.60, 141.87, 137.76, 130.76 (q, J = 34.7), 129.98, 129.33 (q, J = 276.2), 129.20, 127.70, 124.72 (m), 121.98, 119.54, 118.11 (m), 110.55. HRMS(+) calc. for C32H19F12N4O2+: 719.1311 found: 719.1316
3.8. General Procedure for 1H-NMR Titrations
1H-NMR titrations: All titrations were carried out at a 1 mM host concentration in DMSO-d6 with 0.5% H
2O. Stock solutions of guests were prepared using the host solution to ensure that no dilution of the host took place over the course of the titration. TBA salts were dried under a high vacuum prior to the preparation of stock solutions with concentrations in the range of 20–200 mM depending on the titration. Aliquots of guest stock solution were added sequentially to an NMR tube with the free host and a proton spectrum acquired for each addition on a 400 MHz Varian spectrometer. For the chloride titration, the chemical shift of NH protons and other relevant signals were fitted to a 1:2 binding model using Bindfit [
24,
25,
26].
3.9. Molecular Modeling
Modeling was performed by Monte Carlo multiple minimum (MCMM) conformational searches (100 steps per torsion angle, maximum 1000 steps in total), with an energy window of 21 kJ/mol performed in Schrödinger Release 2019-1 using MacroModel in Maestro (version 11.9.011). The force field used is OPLS3e, OPLS_2005, or MMFF, and no implicit solvation. The structures shown are the lowest energy conformations obtained from the calculations.
4. Conclusions
In this work, we have presented the facile synthesis of the novel anion receptor 1,1′-(phenanthrene-3,6-diyl)bis(3-(3,5-bis(trifluoromethyl)phenyl)urea)
13 and shown that
13 interacts with anions of different sizes. The synthetic route presented consists of seven synthetic steps using readily available starting materials and reagents, with only a single step requiring chromatographic separation. Full characterization by
1H and
13C NMR, IR spectroscopy, and mass spectrometry of the intermediates are reported here, which were absent in the previous reports of 3,6-dinitrophenanthrene [
19] and 3,6-diaminophenanthrene [
18]. In addition, the facile synthetic route to the 3,6-diaminophenanthrene (
11) scaffold reported here will lower the barrier for its use in new supramolecular receptors, optoelectronic materials, or other applications. In these applications, the fluorescence of phenanthrene building block
11 and its derivatives could be exploited as well.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}