
Chloro(η2,η2-cycloocta-1,5-diene){1-[(2-[(S)-1-(hydroxymethyl)-3-methylbutyl]amino)-2-oxoethyl]-3-(1-naphthalenylmethyl)benzimidazol-2-ylidene}rhodium(I)


Abstract
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Chloro(η2,η2-Cycloocta-1,5-Diene){1-[(2-[(S)-1-(Hydroxymethyl)-3-Methylbutyl]Amino)-2-Oxoethyl]-3-(1-Naphthalenylmethyl)-Benzimidazol-2-Ylidene}Rhodium(I) (3)
3.2. Iodo(η2,η2-Cycloocta-1,5-Diene)(1,3-Dimethylbenzimidazol-2-Ylidene)Rhodium(I) (4)
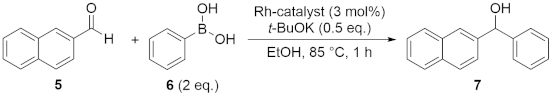
3.3. General Procedure for the Catalytic Reaction of 2-Naphthaldehyde (5) with Phenylboronic Acid (6)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Arduengo, A.J.; Harlow, R.L.; Kline, M. A Stable Crystalline Carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Mukherjee, N.; Mondal, B.; Saha, T.N.; Maity, R. Palladium, iridium, and rhodium complexes bearing chiral N-heterocyclic carbene ligands applied in asymmetric catalysis. Appl. Organomet. Chem. 2022. [Google Scholar] [CrossRef]
- Bellotti, P.; Koy, M.; Hopkinson, M.N.; Glorius, F. Recent advances in the chemistry and applications of N-heterocyclic carbenes. Nature Rev. Chem. 2021, 5, 711–725. [Google Scholar] [CrossRef] [PubMed]
- Nahra, F.; Nelson, D.J.; Nolan, S.P. Design Concepts for N-Heterocyclic Carbene Ligands. Trends Chem. 2020, 2, 1096–1111. [Google Scholar] [CrossRef]
- Peris, E. Smart N-Heterocyclic Carbene Ligands in Catalysis. Chem. Rev. 2018, 118, 9988–10031. [Google Scholar] [CrossRef] [PubMed]
- Janssen-Müller, D.; Schlepphorst, C.; Glorius, F. Privileged chiral N-heterocyclic carbene ligands for asymmetric transition-metal catalysis. Chem. Soc. Rev. 2017, 46, 4845–4854. [Google Scholar] [CrossRef]
- Van Vuuren, E.; Malan, F.P.; Landman, M. Multidentate NHC complexes of group IX metals featuring carbon-based tethers: Synthesis and applications. Coord. Chem. Rev. 2021, 430, 213731. [Google Scholar] [CrossRef]
- Lee, J.; Hahm, H.; Kwak, J.; Kim, M. New Aspects of Recently Developed Rhodium(N-Heterocyclic Carbene)-Catalyzed Organic Transformations. Adv. Synth. Catal. 2019, 361, 1479–1499. [Google Scholar] [CrossRef]
- Peris, E. Routes to N-Heterocyclic Carbene Complexes. In N-Heterocyclic Carbenes in Transition Metal Catalysis. Topics in Organometallic Chemistry; Glorius, F., Ed.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 21, pp. 83–116. [Google Scholar]
- Herrmann, W.A.; Schütz, J.; Frey, G.D.; Herdtweck, E. N-Heterocyclic Carbenes: Synthesis, Structures, and Electronic Ligand Properties. Organometallics 2006, 25, 2437–2448. [Google Scholar] [CrossRef]
- Nelson, D. Accessible Syntheses of Late Transition Metal (Pre)Catalysts Bearing N-Heterocyclic Carbene Ligands. Eur. J. Inorg. Chem. 2015, 2015, 2012–2027. [Google Scholar] [CrossRef]
- Bittermann, A.; Baskakov, D.; Herrmann, W.A. Carbene Complexes Made Easily: Decomposition of Reissert Compounds and Further Synthetic Approaches. Organometallics 2009, 28, 5107–5111. [Google Scholar] [CrossRef]
- Wang, H.M.J.; Lin, I.J.B. Facile Synthesis of Silver(I)-Carbene Complexes. Useful Carbene Transfer Agents. Organometallics 1998, 17, 972–975. [Google Scholar] [CrossRef]
- Lin, J.C.Y.; Huang, R.T.W.; Lee, C.S.; Bhattacharyya, A.; Hwang, W.S.; Lin, I.J.B. Coinage Metal–N-Heterocyclic Carbene Complexes. Chem. Rev. 2009, 109, 3561–3598. [Google Scholar]
- Sakaguchi, S.; Nagao, C.; Ichihara, R.; Matsuo, S. Operationally simple enantioselective silane reduction of ketones by the [Ir(OMe)(cod)]2/azolium catalytic system. Int. J. Org. Chem. 2024, 14, 1–19. [Google Scholar] [CrossRef]
- Enders, D.; Gielen, H.; Runsink, J.; Breuer, K.; Brode, S.; Boehn, K. Diastereoselective Synthesis of Chiral (Triazolinylidene)rhodium Complexes Containing an Axis of Chirality. Eur. J. Inorg. Chem. 1998, 1998, 913–919. [Google Scholar] [CrossRef]
- Oehninger, L.; Küster, L.N.; Schmidt, C.; Muñoz-Castro, A.; Prokop, A.; Ott, I. A Chemical–Biological Evaluation of Rhodium(I) N-Heterocyclic Carbene Complexes as Prospective Anticancer Drugs. Chem. Eur. J. 2013, 19, 17871–17880. [Google Scholar] [CrossRef] [PubMed]
- Köcher, C.; Herrmann, W.A. Heterocyclic carbenes. One-pot synthesis of rhodium and iridium carbene complexes. J. Organomet. Chem. 1997, 532, 261–265. [Google Scholar] [CrossRef]
- He, W.; Zhou, B.; Li, J.; Shi, J. Synthesis of New Chiral Benzimidazolylidene–Rh Complexes and Their Application in Asymmetric Addition Reactions of Organoboronic Acids to Aldehydes. Catalysts 2016, 6, 132. [Google Scholar] [CrossRef]


{kind=link}
 | ||
|---|---|---|
| Entry | Catalyst | Yield (%) 2 |
| 1 | [RhCl(cod)(NHC)] (3) | 79 |
| 2 | [RhI(cod)(NHC)] (4) | 88 |
| 3 | [RhCl(cod)]2 (8) | 14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakaguchi, S.; Matsuo, S. Chloro(η2,η2-cycloocta-1,5-diene){1-[(2-[(S)-1-(hydroxymethyl)-3-methylbutyl]amino)-2-oxoethyl]-3-(1-naphthalenylmethyl)benzimidazol-2-ylidene}rhodium(I). Molbank 2024, 2024, M1810. https://doi.org/10.3390/M1810
Sakaguchi S, Matsuo S. Chloro(η2,η2-cycloocta-1,5-diene){1-[(2-[(S)-1-(hydroxymethyl)-3-methylbutyl]amino)-2-oxoethyl]-3-(1-naphthalenylmethyl)benzimidazol-2-ylidene}rhodium(I). Molbank. 2024; 2024(2):M1810. https://doi.org/10.3390/M1810
Chicago/Turabian StyleSakaguchi, Satoshi, and Shogo Matsuo. 2024. "Chloro(η2,η2-cycloocta-1,5-diene){1-[(2-[(S)-1-(hydroxymethyl)-3-methylbutyl]amino)-2-oxoethyl]-3-(1-naphthalenylmethyl)benzimidazol-2-ylidene}rhodium(I)" Molbank 2024, no. 2: M1810. https://doi.org/10.3390/M1810
APA StyleSakaguchi, S., & Matsuo, S. (2024). Chloro(η2,η2-cycloocta-1,5-diene){1-[(2-[(S)-1-(hydroxymethyl)-3-methylbutyl]amino)-2-oxoethyl]-3-(1-naphthalenylmethyl)benzimidazol-2-ylidene}rhodium(I). Molbank, 2024(2), M1810. https://doi.org/10.3390/M1810