
(2R,4aS,6aS,12bR,14aS,14bR)-N-(2-(2-(2-(2-Azidoethoxy)ethoxy)ethoxy)ethyl)-10-hydroxy-2,4a,6a,9,12b,14a-hexamethyl-11-oxo-1,2,3,4,4a,5,6,6a,11,12b,13,14,14a,14b-tetradecahydropicene-2-carboxamide

 ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
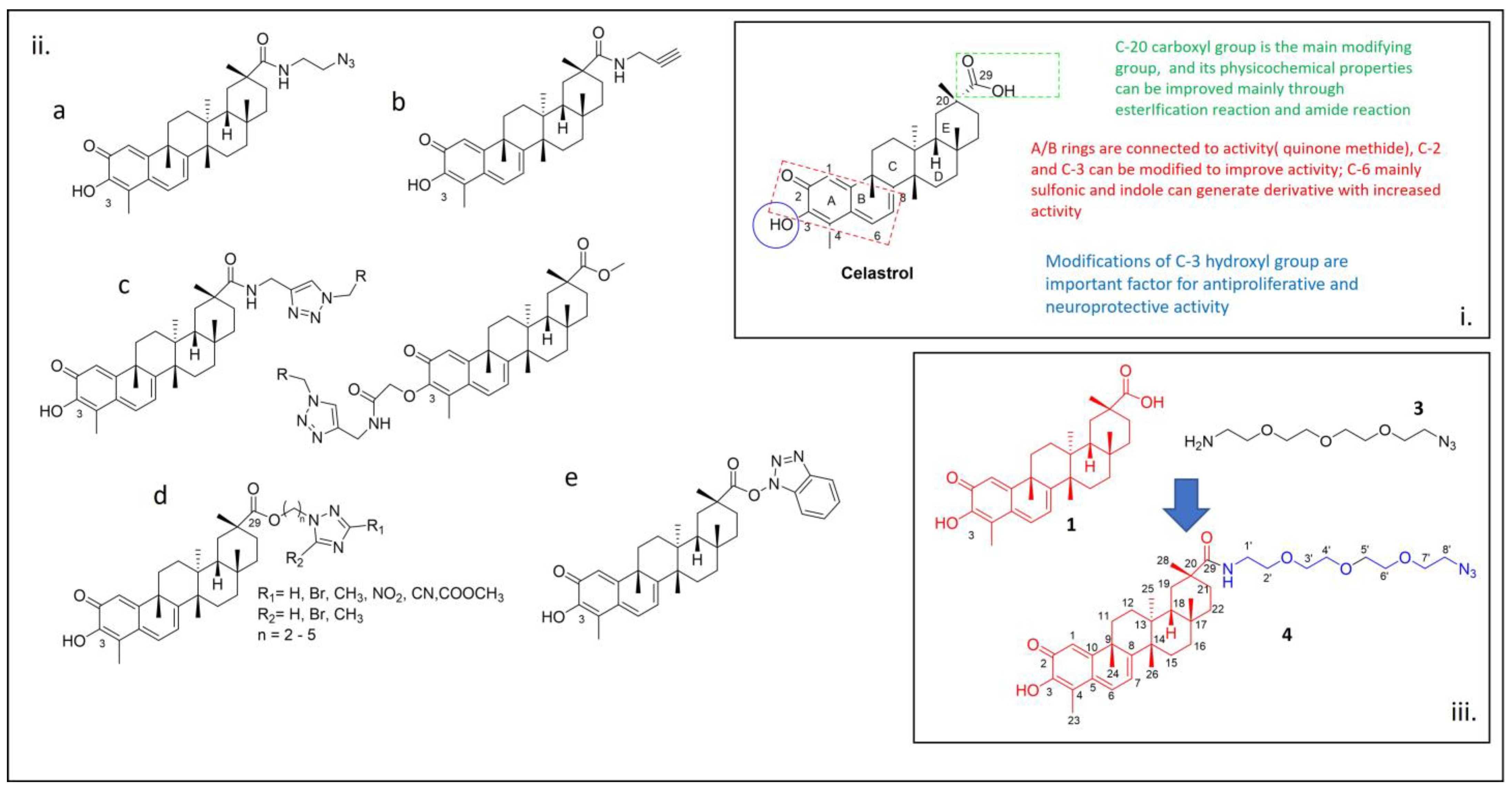
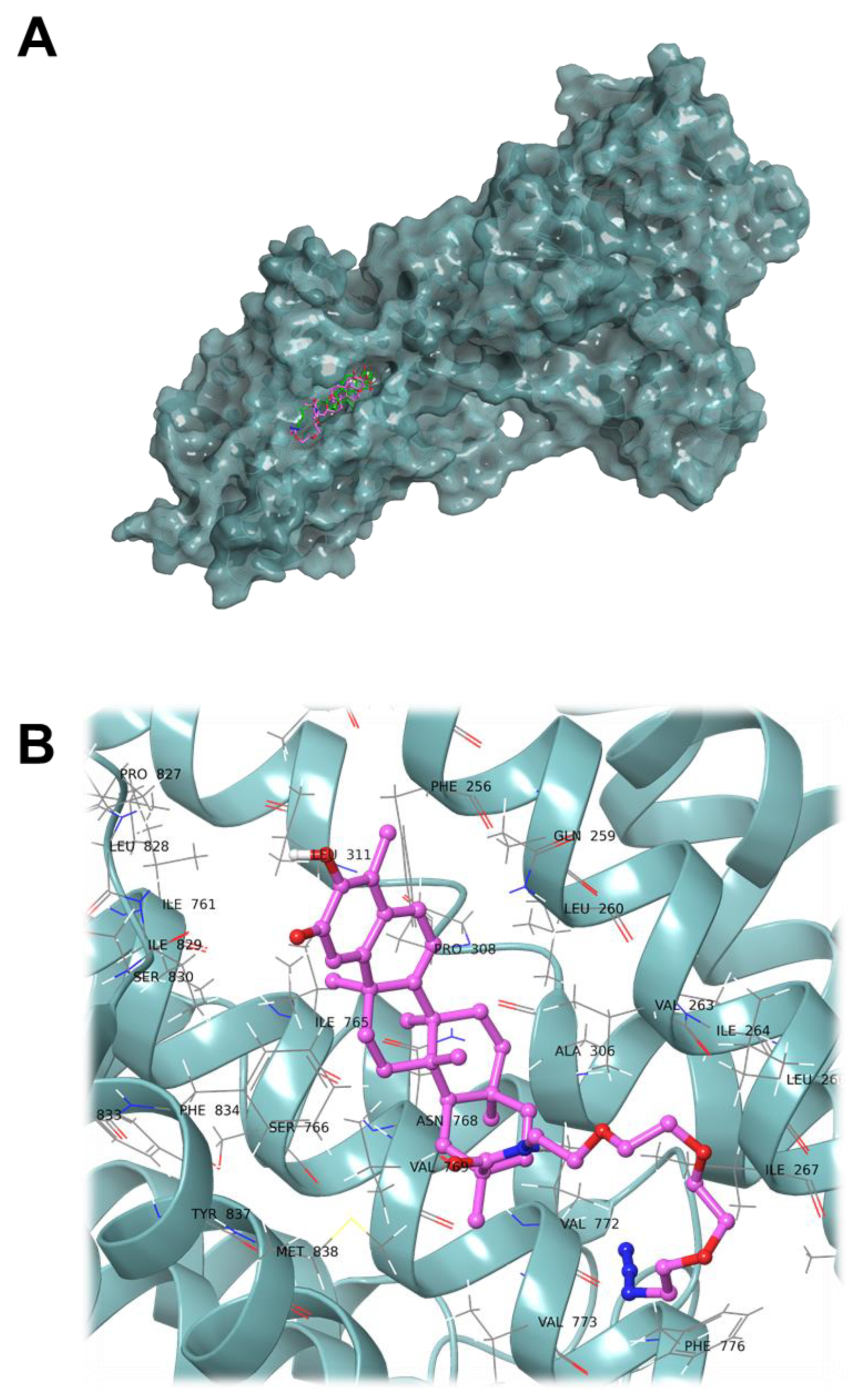
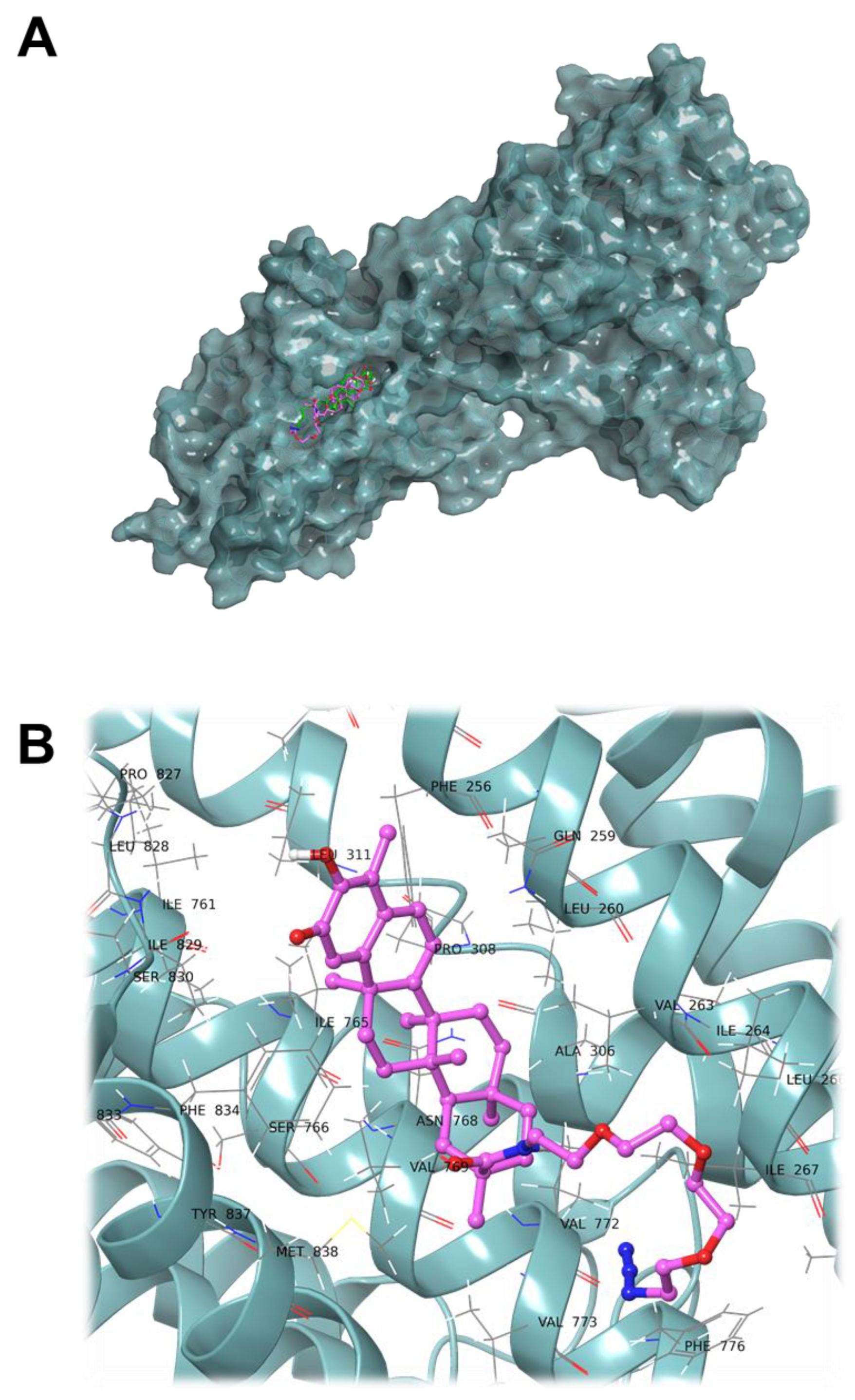
2. Results and Discussion
3. Materials and Methods
3.1. Chemistry
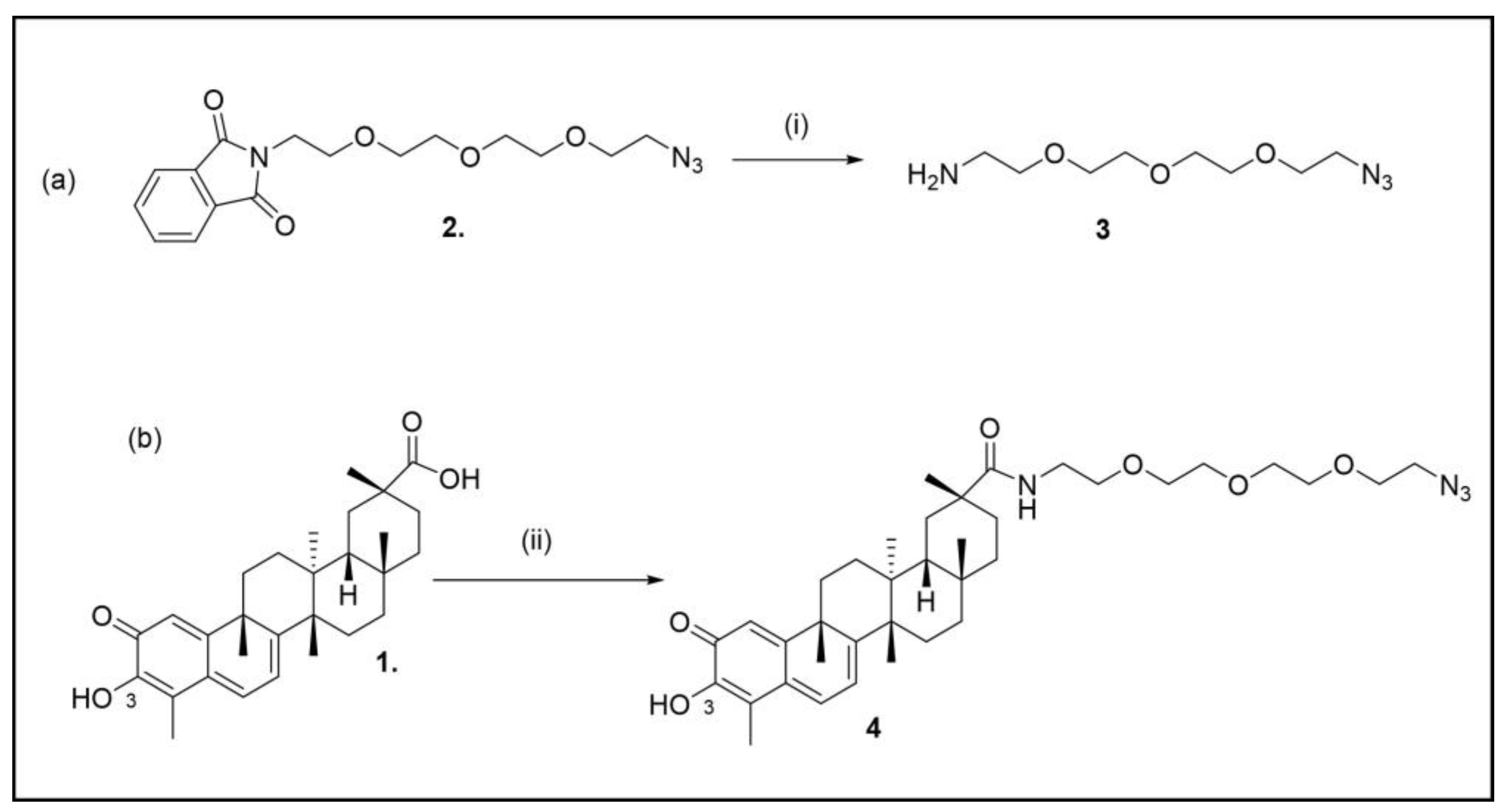
3.1.1. Synthesis of 11-Azido-3,6,9-trioxaundecan-1-amine (3)
3.1.2. Synthesis of (2R,4aS,6aS,12bR,14aS,14bR)10-hydroxy-N-(4-((6-methoxyquinolin-8-yl)amino)pentyl)-2,4a,6a,9,12b,14a-hexamethyl-11-oxo-1,2,3,4,4a,5,6,6a,11,12b,13,14,14a,14b-tetradecahydropicene-2-carboxamide (4)
3.2. Computational Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kannaiyan, R.; Shanmugam, M.K.; Sethi, G. Molecular targets of celastrol derived from Thunder of God Vine: Potential role in the treatment of inflammatory disorders and cancer. Cancer Lett. 2011, 303, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.C.; Cacabelos, R.; Lombardi, V.R.M.; Álvarez, X.A.; Vigo, C. Central Nervous System Effects of Celastrol, a Potent Antioxidant and Antiinflammatory Agent. CNS Drug Rev. 2000, 6, 45–62. [Google Scholar] [CrossRef]
- Der Sarkissian, S.; Cailhier, J.; Borie, M.; Mansour, S.; Hamet, P.; Stevens, L.; Noiseux, N. Celastrol as a Novel Cardioprotective Drug. Can. J. Cardiol. 2013, 29, S331. [Google Scholar] [CrossRef]
- Yang, G.; Wang, K.; Song, H.; Zhu, R.; Ding, S.; Yang, H.; Sun, J.; Wen, X.; Sun, L. Celastrol ameliorates osteoarthritis via regulating TLR2/NF-κB signaling pathway. Front. Pharm. 2022, 13, 963506. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.Y.; Jang, S.-W.; Ko, J. Celastrol inhibits the growth of estrogen positive human breast cancer cells through modulation of estrogen receptor α. Cancer Lett. 2011, 300, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Xu, L.; Yu, P.; Jiang, J.; Zhang, G.; Wang, Y. Synthesis and preliminary evaluation of neuroprotection of celastrol analogues in PC12 cells. Bioorganic Med. Chem. Lett. 2010, 20, 3844–3847. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lee, J.; Salazar Hernandez, M.A.; Mazitschek, R.; Ozcan, U. Treatment of Obesity with Celastrol. Cell 2015, 161, 999–1011. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.Z.; Hwang, B.Y.; Kim, H.S.; Lee, J.H.; Kim, Y.H.; Lee, J.J. Antiinflammatory Constituents of Celastrus orbiculatus Inhibit the NF-κB Activation and NO Production. J. Nat. Prod. 2002, 65, 89–91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Li, Y.; Yu, Y.; Zou, P.; Jiang, Y.; Sun, D. Characterization of Celastrol to Inhibit Hsp90 and Cdc37 Interaction. J. Biol. Chem. 2009, 284, 35381–35389. [Google Scholar] [CrossRef]
- Wong, V.K.W.; Qiu, C.; Xu, S.-W.; Law, B.Y.K.; Zeng, W.; Wang, H.; Michelangeli, F.; Dias, I.R.D.S.R.; Qu, Y.Q.; Chan, T.W.; et al. Ca2+ signalling plays a role in celastrol-mediated suppression of synovial fibroblasts of rheumatoid arthritis patients and experimental arthritis in rats. Br. J. Pharmacol. 2019, 176, 2922–2944. [Google Scholar] [CrossRef]
- Di Matteo, A.; Bathon, J.M.; Emery, P. Rheumatoid arthritis. Lancet 2023, 402, 2019–2033. [Google Scholar] [CrossRef] [PubMed]
- Gravallese, E.M.; Firestein, G.S. Rheumatoid Arthritis–Common Origins, Divergent Mechanisms. N. Engl. J. Med. 2023, 388, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.Y.; Yin, H.X.; Li, S.F.; Chen, Y.; Zhao, Y.J.; Hu, W.; Zhou, R.P. Calcium-Permeable Channels Cooperation for Rheumatoid Arthritis: Therapeutic Opportunities. Biomolecules 2022, 12, 1383. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.K.; Li, T.; Law, B.Y.; Ma, E.D.; Yip, N.C.; Michelangeli, F.; Law, C.K.; Zhang, M.M.; Lam, K.Y.; Chan, P.L.; et al. Saikosaponin-d, a novel SERCA inhibitor, induces autophagic cell death in apoptosis-defective cells. Cell Death Dis. 2013, 4, e720. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.-W.; Law, B.Y.K.; Qu, S.L.Q.; Hamdoun, S.; Chen, J.; Zhang, W.; Guo, J.-R.; Wu, A.-G.; Mok, S.W.F.; Zhang, D.W.; et al. SERCA and P-glycoprotein inhibition and ATP depletion are necessary for celastrol-induced autophagic cell death and collateral sensitivity in multidrug-resistant tumor cells. Pharmacol. Res. 2020, 153, 104660. [Google Scholar] [CrossRef]
- Lv, Q.W.; Zhang, W.; Shi, Q.; Zheng, W.J.; Li, X.; Chen, H.; Wu, Q.J.; Jiang, W.L.; Li, H.B.; Gong, L.; et al. Comparison of Tripterygium wilfordii Hook F with methotrexate in the treatment of active rheumatoid arthritis (TRIFRA): A randomised, controlled clinical trial. Ann. Rheum. Dis. 2015, 74, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Klaić, L.; Morimoto, R.I.; Silverman, R.B. Celastrol Analogues as Inducers of the Heat Shock Response. Design and Synthesis of Affinity Probes for the Identification of Protein Targets. ACS Chem. Biol. 2012, 7, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Coghi, P.; Ng, J.P.L.; Kadioglu, O.; Law, B.Y.K.; Qiu, A.C.; Saeed, M.E.M.; Chen, X.; Ip, C.K.; Efferth, T.; Liu, L.; et al. Synthesis, computational docking and biological evaluation of celastrol derivatives as dual inhibitors of SERCA and P-glycoprotein in cancer therapy. Eur. J. Med. Chem. 2021, 224, 113676. [Google Scholar] [CrossRef]
- Xie, Y.; Kuan, H.; Wei, Q.; Gianoncelli, A.; Ribaudo, G.; Coghi, P. (2R,4aS,6aS,12bR,14aS,14bR)10-Hydroxy-N-(4-((6-methoxyquinolin-8-yl)amino)pentyl)-2,4a,6a,9,12b,14a-hexamethyl-11-oxo-1,2,3,4,4a,5,6,6a,11,12b,13,14,14a,14b-tetradecahydropicene-2-carboxamide. Molbank 2023, 2023, M1716. [Google Scholar] [CrossRef]
- Nasra, S.; Bhatia, D.; Kumar, A. Recent advances in nanoparticle-based drug delivery systems for rheumatoid arthritis treatment. Nanoscale Adv. 2022, 4, 3479–3494. [Google Scholar] [CrossRef]
- Crescenzi, V.; Cornelio, L.; Di Meo, C.; Nardecchia, S.; Lamanna, R. Novel hydrogels via click chemistry: Synthesis and potential biomedical applications. Biomacromolecules 2007, 8, 1844–1850. [Google Scholar] [CrossRef]
- Saxon, E.; Armstrong, J.I.; Bertozzi, C.R. A “traceless” Staudinger ligation for the chemoselective synthesis of amide bonds. Org. Lett. 2000, 2, 2141–2143. [Google Scholar] [CrossRef] [PubMed]
- Saxon, E.; Bertozzi, C.R. Cell surface engineering by a modified Staudinger reaction. Science 2000, 287, 2007–2010. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Liu, D.; Zhang, Q.; Yang, F.; Wong, Y.K.; Xia, F.; Zhang, J.; Chen, J.; Tian, Y.; Yang, C.; et al. Celastrol induces ferroptosis in activated HSCs to ameliorate hepatic fibrosis via targeting peroxiredoxins and HO-1. Acta Pharm. Sin. B 2022, 12, 2300–2314. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Wang, W.; Zhang, Y.; Fu, X.; Ping, K.; Zhao, J.; Lei, Y.; Mou, Y.; Wang, S. Synthesis and biological evaluation of celastrol derivatives as potential anti-glioma agents by activating RIP1/RIP3/MLKL pathway to induce necroptosis. Eur. J. Med. Chem. 2022, 229, 114070. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Chen, C.; Zhu, H.; Shi, Z.; Sun, J.; Chen, L. Discovery of novel celastrol-triazole derivatives with Hsp90-Cdc37 disruption to induce tumor cell apoptosis. Bioorg. Chem. 2021, 111, 104867. [Google Scholar] [CrossRef] [PubMed]
- He, Q.-W.; Feng, J.-H.; Hu, X.-L.; Long, H.; Huang, X.-F.; Jiang, Z.-Z.; Zhang, X.-Q.; Ye, W.-C.; Wang, H. Synthesis and Biological Evaluation of Celastrol Derivatives as Potential Immunosuppressive Agents. J. Nat. Prod. 2020, 83, 2578–2586. [Google Scholar] [CrossRef] [PubMed]
- Axer, A.; Hermann, S.; Kehr, G.; Clases, D.; Karst, U.; Fischer-Riepe, L.; Roth, J.; Fobker, M.; Schäfers, M.; Gilmour, R.; et al. Harnessing the Maltodextrin Transport Mechanism for Targeted Bacterial Imaging: Structural Requirements for Improved in vivo Stability in Tracer Design. ChemMedChem 2018, 13, 241–250. [Google Scholar] [CrossRef]
- Pang, C.; Luo, J.; Liu, C.; Wu, X.; Wang, D. Synthesis and Biological Evaluation of a Series of Novel Celastrol Derivatives with Amino Acid Chain. Chem. Biodivers. 2018, 15, e1800059. [Google Scholar] [CrossRef]
- Toyoshima, C.; Nomura, H. Structural changes in the calcium pump accompanying the dissociation of calcium. Nature 2002, 418, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuzhu, G.; Anyanwu, M.; Yang, X.; Zimo, R.; Gianoncelli, A.; Ribaudo, G.; Coghi, P. (2R,4aS,6aS,12bR,14aS,14bR)-N-(2-(2-(2-(2-Azidoethoxy)ethoxy)ethoxy)ethyl)-10-hydroxy-2,4a,6a,9,12b,14a-hexamethyl-11-oxo-1,2,3,4,4a,5,6,6a,11,12b,13,14,14a,14b-tetradecahydropicene-2-carboxamide. Molbank 2024, 2024, M1800. https://doi.org/10.3390/M1800
Yuzhu G, Anyanwu M, Yang X, Zimo R, Gianoncelli A, Ribaudo G, Coghi P. (2R,4aS,6aS,12bR,14aS,14bR)-N-(2-(2-(2-(2-Azidoethoxy)ethoxy)ethoxy)ethyl)-10-hydroxy-2,4a,6a,9,12b,14a-hexamethyl-11-oxo-1,2,3,4,4a,5,6,6a,11,12b,13,14,14a,14b-tetradecahydropicene-2-carboxamide. Molbank. 2024; 2024(2):M1800. https://doi.org/10.3390/M1800
Chicago/Turabian StyleYuzhu, Guo, Margrate Anyanwu, Xiao Yang, Ren Zimo, Alessandra Gianoncelli, Giovanni Ribaudo, and Paolo Coghi. 2024. "(2R,4aS,6aS,12bR,14aS,14bR)-N-(2-(2-(2-(2-Azidoethoxy)ethoxy)ethoxy)ethyl)-10-hydroxy-2,4a,6a,9,12b,14a-hexamethyl-11-oxo-1,2,3,4,4a,5,6,6a,11,12b,13,14,14a,14b-tetradecahydropicene-2-carboxamide" Molbank 2024, no. 2: M1800. https://doi.org/10.3390/M1800
APA StyleYuzhu, G., Anyanwu, M., Yang, X., Zimo, R., Gianoncelli, A., Ribaudo, G., & Coghi, P. (2024). (2R,4aS,6aS,12bR,14aS,14bR)-N-(2-(2-(2-(2-Azidoethoxy)ethoxy)ethoxy)ethyl)-10-hydroxy-2,4a,6a,9,12b,14a-hexamethyl-11-oxo-1,2,3,4,4a,5,6,6a,11,12b,13,14,14a,14b-tetradecahydropicene-2-carboxamide. Molbank, 2024(2), M1800. https://doi.org/10.3390/M1800