
Butyl (2,2-Dibutoxybutanoyl)-ʟ-Tryptophanate



Abstract
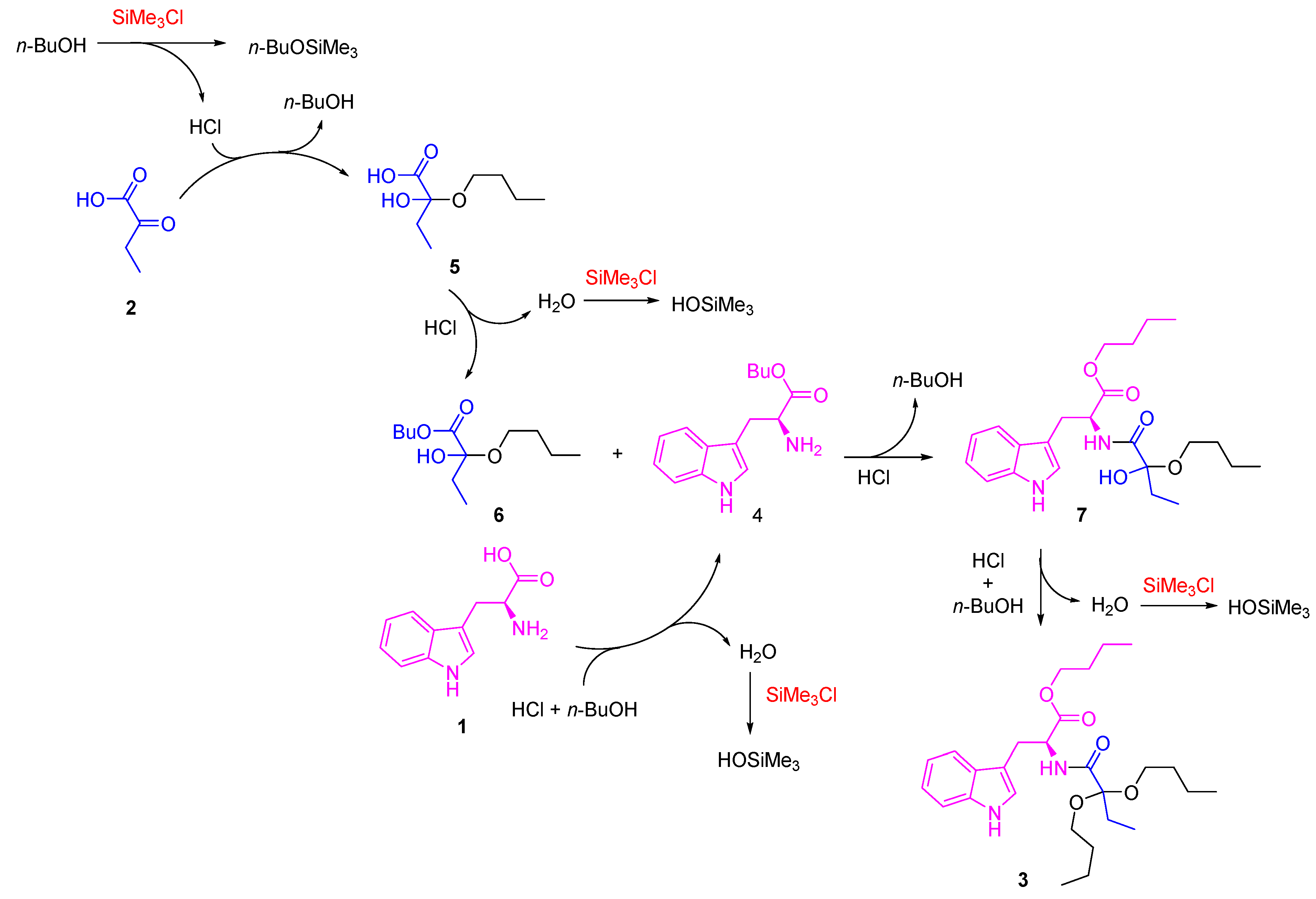
1. Introduction
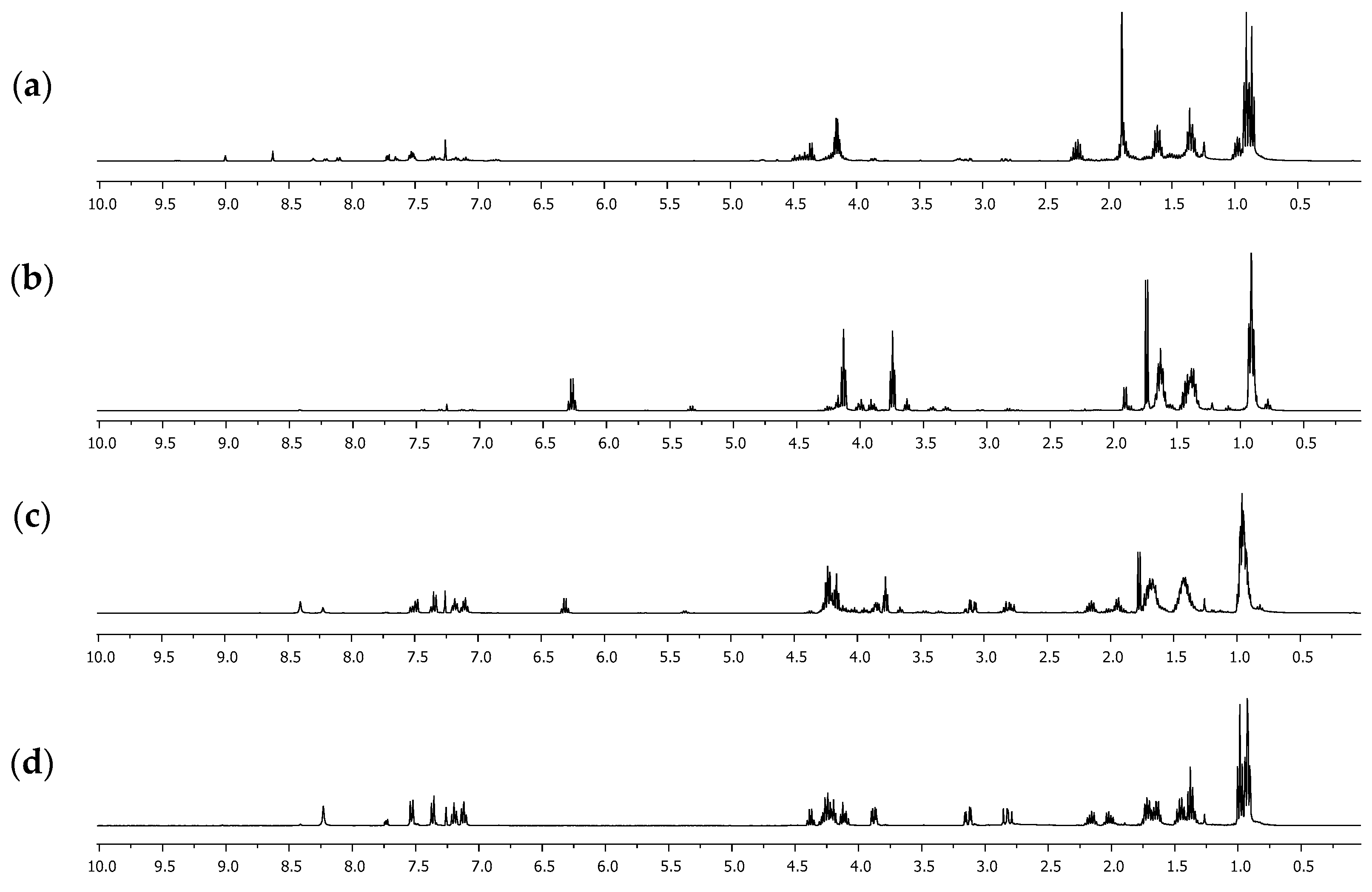
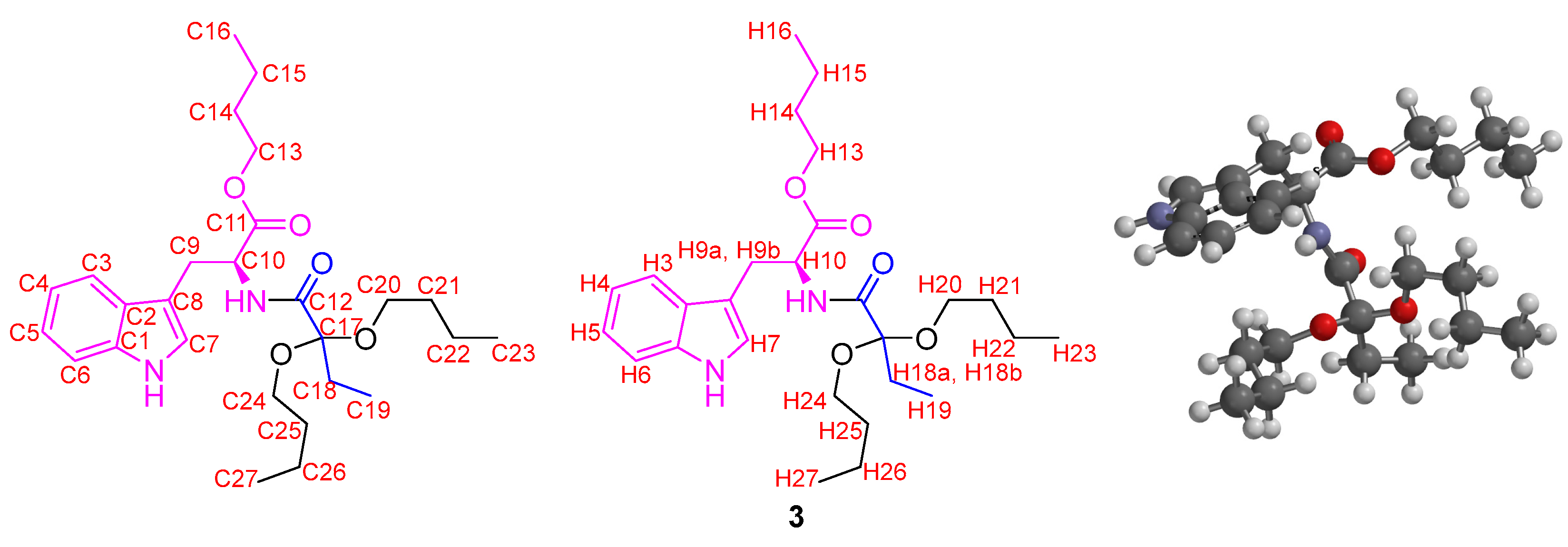
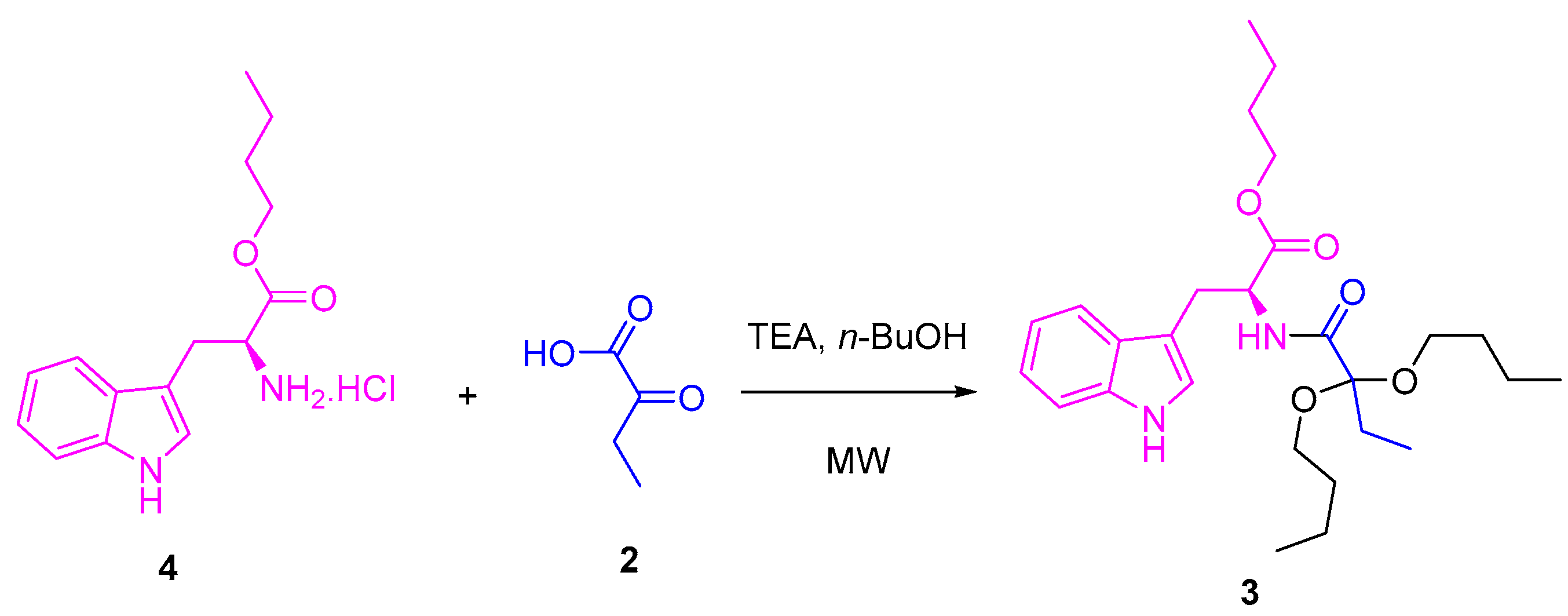
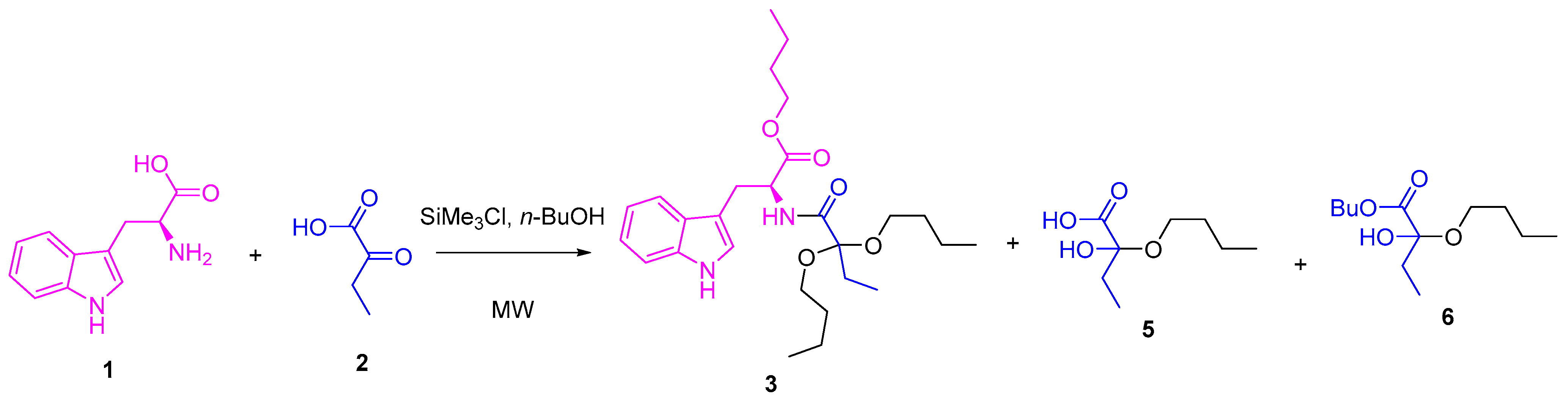
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Chemical Synthesis
3.3. DFT B3LYP Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, D.; Porter, W.R.; Zhang, G.G.Z. Drug Stability and Degradation Studies. In Developing Solid Oral Dosage Forms; Elsevier: Amsterdam, The Netherlands, 2017; pp. 113–149. ISBN 978-0-12-802447-8. [Google Scholar]
- Grindley, T.B.; Gulasekharam, V. Benzylidene Acetal Structural Elucidation by N.M.R. Spectroscopy: Application of Carbon-13. N.M.R.-Spectral Parameters. Carbohydr. Res. 1979, 74, 7–30. [Google Scholar] [CrossRef]
- Orliac, A.; Gomez Pardo, D.; Bombrun, A.; Cossy, J. XtalFluor-E, an Efficient Coupling Reagent for Amidation of Carboxylic Acids. Org. Lett. 2013, 15, 902–905. [Google Scholar] [CrossRef]
- Jereb, M.; Vražič, D.; Zupan, M. Iodine-Catalyzed Transformation of Molecules Containing Oxygen Functional Groups. Tetrahedron 2011, 67, 1355–1387. [Google Scholar] [CrossRef]
- Huang, J.; Lu, Y.; Qiu, B.; Liang, Y.; Li, N.; Dong, D. One-Pot Synthesis of Substituted Isothiazol-3(2 H)-Ones: Intramolecular Annulation of α-Carbamoyl Ketene-S,S-Acetals via PIFA-Mediated N-S Bond Formation. Synthesis 2007, 2007, 2791–2796. [Google Scholar] [CrossRef]
- Sheng, G.; Zhang, W. New Advances of the Methods of Amide Function Group for Construction. Chin. J. Org. Chem. 2013, 33, 2271. [Google Scholar] [CrossRef][Green Version]
- Masse, C.E.; Yang, M.; Solomon, J.; Panek, J.S. Total Synthesis of (+)-Mycotrienol and (+)-Mycotrienin I: Application of Asymmetric Crotylsilane Bond Constructions. J. Am. Chem. Soc. 1998, 120, 4123–4134. [Google Scholar] [CrossRef]
- Kidjemet, D. N,N-Dimethylformamide Dimethyl Acetal. Synlett 2002, 2002, 1741–1742. [Google Scholar] [CrossRef]
- Kinderman, S.S.; de Gelder, R.; van Maarseveen, J.H.; Schoemaker, H.E.; Hiemstra, H.; Rutjes, F.P.J.T. Amidopalladation of Alkoxyallenes Applied in the Synthesis of an Enantiopure 1-Ethylquinolizidine Frog Alkaloid. J. Am. Chem. Soc. 2004, 126, 4100–4101. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, A. Cerium(IV) Ammonium Nitrate: A Versatile Oxidant in Synthetic Organic Chemistry. Synlett 2005, 2005, 3014–3015. [Google Scholar] [CrossRef]
- Lippur, K.; Kanger, T.; Kriis, K.; Kailas, T.; Müürisepp, A.-M.; Pehk, T.; Lopp, M. Synthesis of (2S,2′S)-Bimorpholine N,N′-Quaternary Salts as Chiral Phase Transfer Catalysts. Tetrahedron Asymmetry 2007, 18, 137–141. [Google Scholar] [CrossRef]
- Juma, B.; Adeel, M.; Villinger, A.; Langer, P. Efficient Synthesis of 2,6-Dioxo-1,2,3,4,5,6-Hexahydroindoles Based on the Synthesis and Reactions of (2,4-Dioxocyclohex-1-Yl)Acetic Acid Derivatives. Tetrahedron Lett. 2008, 49, 2272–2274. [Google Scholar] [CrossRef]
- Carmali, S.; Brocchini, S. Polyacetals. In Natural and Synthetic Biomedical Polymers; Elsevier: Amsterdam, The Netherlands, 2014; pp. 219–233. ISBN 978-0-12-396983-5. [Google Scholar]
- Yanev, P.; Angelov, P. Synthesis of Functionalised β-Keto Amides by Aminoacylation/Domino Fragmentation of β-Enamino Amides. Beilstein J. Org. Chem. 2018, 14, 2602–2606. [Google Scholar] [CrossRef] [PubMed]
- Zaher, S.; Christ, L.; Abd El Rahim, M.; Kanj, A.; Karamé, I. Green Acetalization of Glycerol and Carbonyl Catalyzed by FeCl3·6H2O. Mol. Catal. 2017, 438, 204–213. [Google Scholar] [CrossRef]
- Halli, J.; Hofman, K.; Beisel, T.; Manolikakes, G. Synthesis of N-Acyl-N,O-acetals from Aldehydes, Amides and Alcohols. Eur. J. Org. Chem. 2015, 2015, 4624–4627. [Google Scholar] [CrossRef]
- Sattenapally, N.; Sharma, J.; Hou, Y. Selective Conversion of Primary Amides to Esters Promoted by KHSO4. Arkivoc 2018, 2018, 174–183. [Google Scholar] [CrossRef]
- Miranda, L.P.; Jones, A.; Meutermans, W.D.F.; Alewood, P.F. P-Cresol As a Reversible Acylium Ion Scavenger in Solid-Phase Peptide Synthesis. J. Am. Chem. Soc. 1998, 120, 1410–1420. [Google Scholar] [CrossRef]
- Vincent, S.; Lebeau, L.; Mioskowski, C. N, N-Dibenzyl Formamide Dimethyl Acetal and N, N-Dibenzyl Chloromethylene Iminium Chloride: Two Complementary Reagents for the Protection Of Primary Amines as N, N-Dibenzyl Formamidines. Synth. Commun. 1999, 29, 167–174. [Google Scholar] [CrossRef]
- Tsotinis, A.; Vlachou, M.; Kiakos, K.; Hartley, J.A.; Thurston, D.E. Design and Synthesis of Two Cytotoxic Analogs of the Novel Pyrrolo[1′,2′:1,2][1,4]Diazepin [7,6-b]Indol-5(6H)-One Nucleus. Chem. Lett. 2003, 32, 512–513. [Google Scholar] [CrossRef]
- Wu, Y.-C.; Li, H.-J.; Liu, L.; Demoulin, N.; Liu, Z.; Wang, D.; Chen, Y.-J. Facile Synthesis of Spiropyrans from Chromene Hemiacetal Esters and Bifunctional Nucleophiles. Synlett 2011, 2011, 1573–1578. [Google Scholar] [CrossRef]
- Luo, C.; Xu, Q.; Huang, C.; Luo, L.; Zhu, J.; Zhang, R.; Huang, G.; Yin, D. Development of an Efficient Synthetic Process for Broflanilide. Org. Process Res. Dev. 2020, 24, 1024–1031. [Google Scholar] [CrossRef]
- Quiroga, D. Employing Molecular Docking Calculations for the Design of Alkyl (2-Alcoxy-2-Hydroxypropanoyl)-ʟ-Tryptophanate Derivatives as Potential Inhibitors of 11β-Hydroxysteroid Dehydrogenase Type 1 (11β-HSD1). Reactions 2023, 4, 108–116. [Google Scholar] [CrossRef]
- Ma, X.-Y.; Shao, F.-Q.; Hu, X.; Liu, X. Progress in the Synthesis of N-Acyl-N,O-Acetals. Synthesis 2022, 54, 1203–1216. [Google Scholar] [CrossRef]
- Iwai, K.; Hikasa, A.; Yoshioka, K.; Tani, S.; Umezu, K.; Nishiwaki, N. Synthesis of Bis(Functionalized) Aminals via Successive Nucleophilic Amidation and Amination. J. Org. Chem. 2023, 88, 2207–2213. [Google Scholar] [CrossRef]
- Quiroga, D.; Becerra, L.; Sadat-Bernal, J.; Vargas, N.; Coy-Barrera, E. Synthesis and Antifungal Activity against Fusarium Oxysporum of Some Brassinin Analogs Derived from ʟ-Tryptophan: A DFT/B3LYP Study on the Reaction Mechanism. Molecules 2016, 21, 1349. [Google Scholar] [CrossRef]
- Liu, B.; Thayumanavan, S. Substituent Effects on the pH Sensitivity of Acetals and Ketals and Their Correlation with Encapsulation Stability in Polymeric Nanogels. J. Am. Chem. Soc. 2017, 139, 2306–2317. [Google Scholar] [CrossRef]
- Phan, T.B.; Mayr, H. Comparison of the Nucleophilicities of Alcohols and Alkoxides. Can. J. Chem. 2005, 83, 1554–1560. [Google Scholar] [CrossRef]
- Zhao, W.; Chen, F.-E. One-Pot Synthesis and Its Practical Application in Pharmaceutical Industry. Curr. Org. Synth. 2012, 9, 873–897. [Google Scholar] [CrossRef]
- Hayashi, Y. Pot Economy and One-Pot Synthesis. Chem. Sci. 2016, 7, 866–880. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Zhang, W. Recent Developments in One-Pot Stepwise Synthesis (OPSS) of Small Molecules. iScience 2022, 25, 105005. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sha, Y. A Convenient Synthesis of Amino Acid Methyl Esters. Molecules 2008, 13, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Izumi, M.; Fukase, K.; Kusumoto, S. TMSCl as a Mild and Effective Source of Acidic Catalysis in Fischer Glycosidation and Use of Propargyl Glycoside for Anomeric Protection. Biosci. Biotechnol. Biochem. 2002, 66, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Ward, D.J.; Saccomando, D.J.; Walker, G.; Mansell, S.M. Sustainable Routes to Alkenes: Applications of Homogeneous Catalysis to the Dehydration of Alcohols to Alkenes. Catal. Sci. Technol. 2023, 13, 2638–2647. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Conditions | Yield of 3 (%) |
|---|---|---|
| 1 | 80 °C, 20 min | No detected |
| 2 | 80 °C, 40 min | No detected |
| 3 | 120 °C, 20 min | 5 |
| 4 | 120 °C, 40 min | 7 |
| 5 | 140 °C, 20 min | 13 |
| 6 | 140 °C, 40 min | 22 |
| Entry | Conditions | Yield of 3 (%) | Yield of 5 (%) | Yield of 6 (%) |
|---|---|---|---|---|
| 1 | 80 °C, 20 min | No detected | 15 | 2 |
| 2 | 80 °C, 40 min | No detected | 31 | 4 |
| 3 | 120 °C, 20 min | No detected | 3 | 23 |
| 4 | 120 °C, 40 min | No detected | No detected | 51 |
| 5 | 140 °C, 20 min | 47 | No detected | 25 |
| 6 | 140 °C, 40 min | 89 | No detected | No detected |
| C | Experimental δC | Calculated δC | ΔδC | H | Experimental δH | Calculated δH | ΔδH |
|---|---|---|---|---|---|---|---|
| C1 | 136.7 | 141.3 | 4.6 | H3 | 7.53 | 7.69 | 0.16 |
| C2 | 132.0 | 134.5 | 2.4 | H4 | 7.37 | 7.37 | 0.00 |
| C3 | 122.35 | 121.9 | 0.5 | H5 | 7.13 | 7.39 | 0.26 |
| C4 | 126.6 | 125.2 | 1.4 | H6 | 7.20 | 7.31 | 0.11 |
| C5 | 129.0 | 127.3 | 1.7 | H7 | 8.23 | 7.00 | 1.23 |
| C6 | 118.6 | 114.3 | 4.3 | H9a | 2.84 | 3.11 | 0.27 |
| C7 | 119.7 | 117.6 | 2.1 | H9b | 3.14 | 3.17 | 0.03 |
| C8 | 131.0 | 126.4 | 4.6 | H10 | 3.88 | 3.32 | 0.56 |
| C9 | 30.7 | 30.0 | 0.7 | H13 | 4.20 | 3.62 | 0.58 |
| C10 | 54.3 | 63.9 | 9.6 | H14 | 1.41 | 2.06 | 0.65 |
| C11 | 174.1 | 177.5 | 3.4 | H15 | 1.41 | 1.23 | 0.18 |
| C12 | 172.9 | 174.2 | 1.3 | H16 | 0.99 | 1.12 | 0.13 |
| C13 | 65.7 | 69.5 | 3.8 | H18a | 2.06 | 1.87 | 0.19 |
| C14 | 30.8 | 34.5 | 3.7 | H18b | 2.01 | 1.37 | 0.64 |
| C15 | 14.2 | 23.9 | 9.7 | H19 | 0.93 | 1.32 | 0.39 |
| C16 | 8.2 | 16.3 | 8.1 | H20 | 4.10 | 4.11 | 0.01 |
| C17 | 110.6 | 106.3 | 4.3 | H21 | 1.41 | 1.97 | 0.56 |
| C18 | 33.7 | 35.4 | 1.7 | H22 | 1.41 | 1.47 | 0.06 |
| C19 | 13.9 | 11.6 | 2.3 | H23 | 0.99 | 1.06 | 0.07 |
| C20 | 63.2 | 65.7 | 2.5 | H24 | 4.27 | 3.74 | 0.53 |
| C21 | 30.7 | 36.9 | 6.2 | H25 | 1.41 | 2.48 | 1.07 |
| C22 | 25.4 | 25.5 | 0.1 | H26 | 1.41 | 1.48 | 0.07 |
| C23 | 19.3 | 16.7 | 2.6 | H27 | 0.99 | 0.85 | 0.14 |
| C24 | 65.2 | 65.8 | 0.6 | ||||
| C25 | 30.7 | 35.4 | 4.7 | ||||
| C26 | 25.4 | 25.1 | 0.3 | ||||
| C27 | 19.3 | 16.0 | 3.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quiroga, D.; Coy-Barrera, E. Butyl (2,2-Dibutoxybutanoyl)-ʟ-Tryptophanate. Molbank 2024, 2024, M1794. https://doi.org/10.3390/M1794
Quiroga D, Coy-Barrera E. Butyl (2,2-Dibutoxybutanoyl)-ʟ-Tryptophanate. Molbank. 2024; 2024(1):M1794. https://doi.org/10.3390/M1794
Chicago/Turabian StyleQuiroga, Diego, and Ericsson Coy-Barrera. 2024. "Butyl (2,2-Dibutoxybutanoyl)-ʟ-Tryptophanate" Molbank 2024, no. 1: M1794. https://doi.org/10.3390/M1794
APA StyleQuiroga, D., & Coy-Barrera, E. (2024). Butyl (2,2-Dibutoxybutanoyl)-ʟ-Tryptophanate. Molbank, 2024(1), M1794. https://doi.org/10.3390/M1794