
Structural Elucidation of the Triethylammonium Betaine of Squaric Acid †

 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
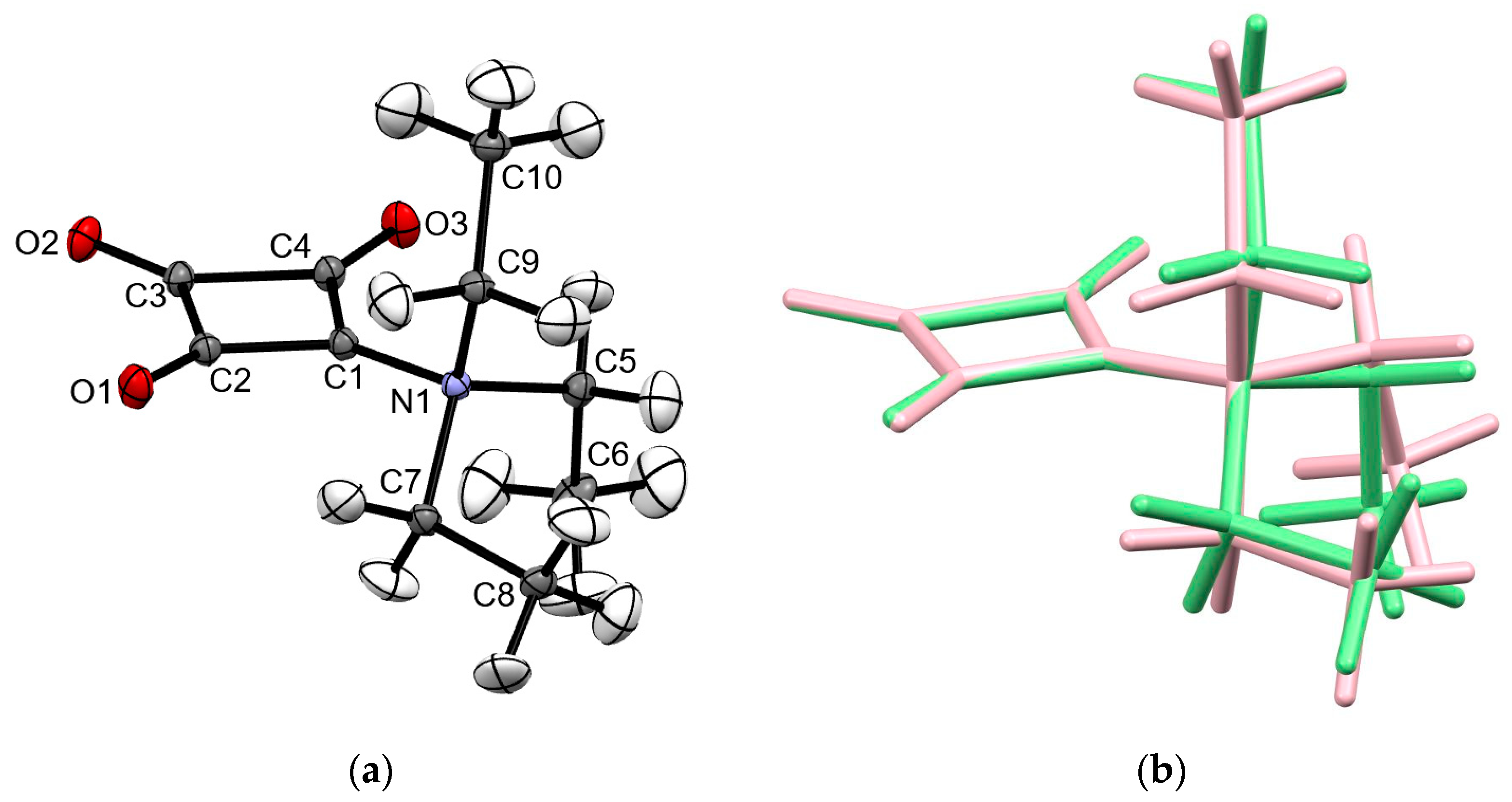
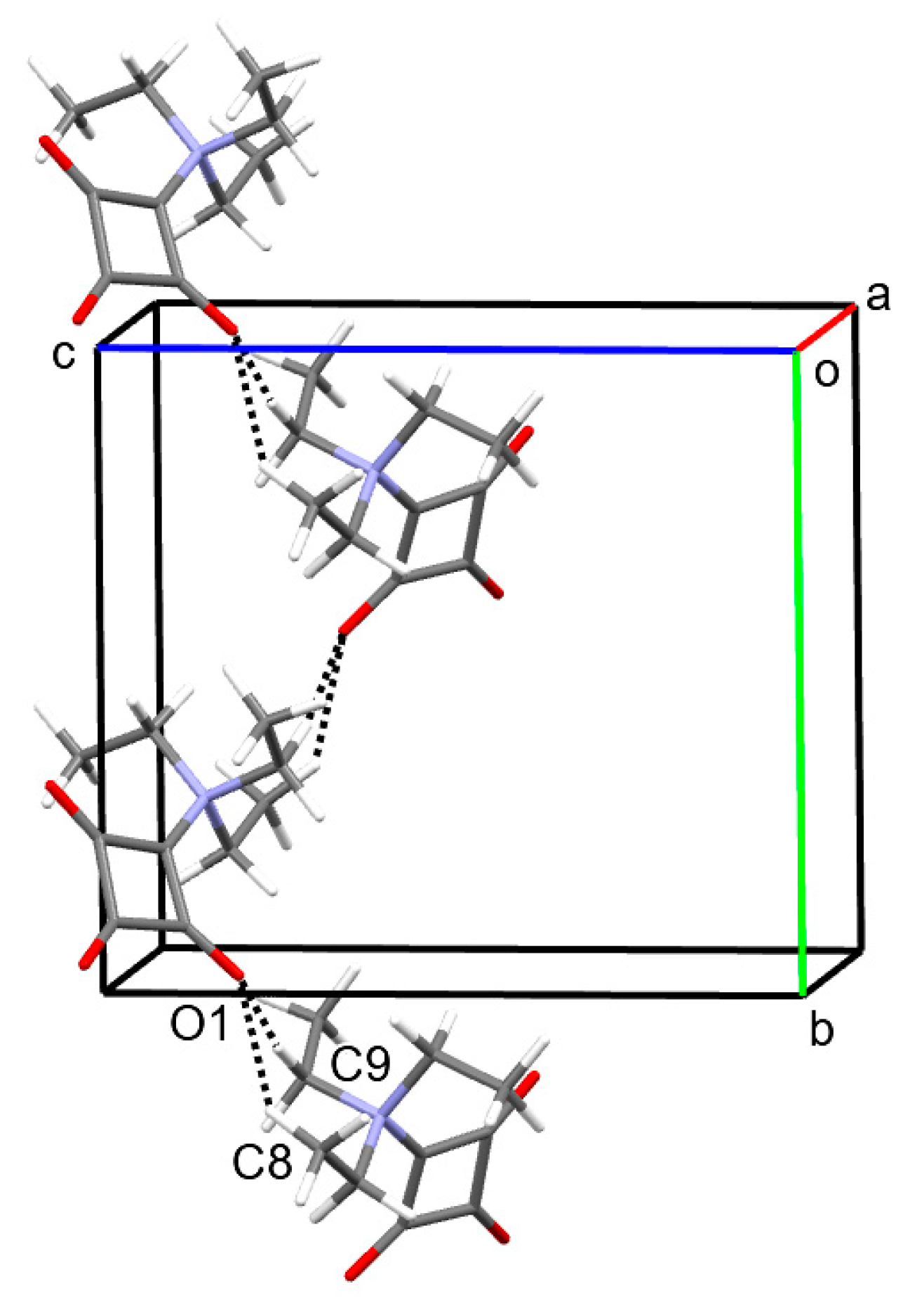
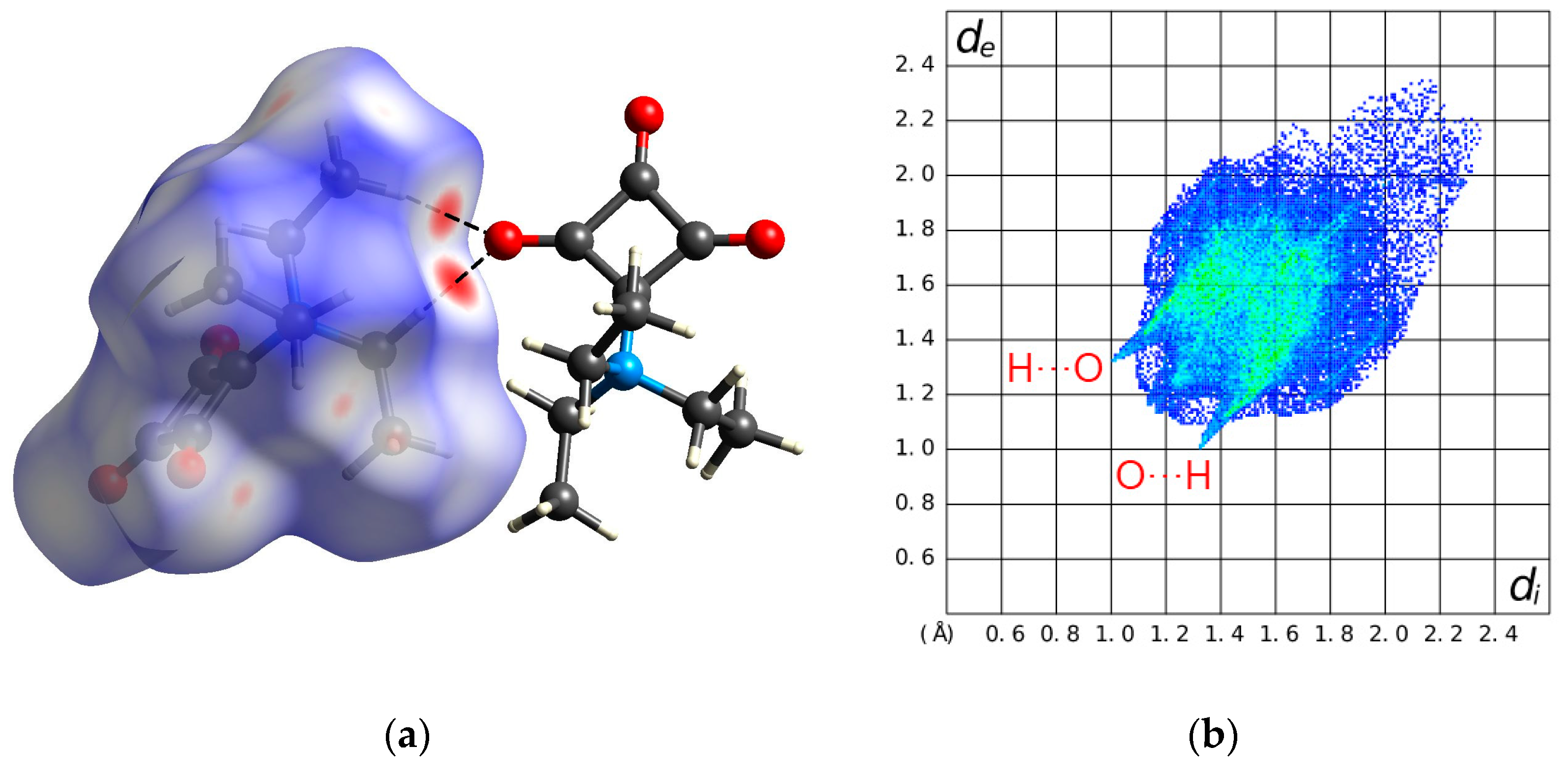
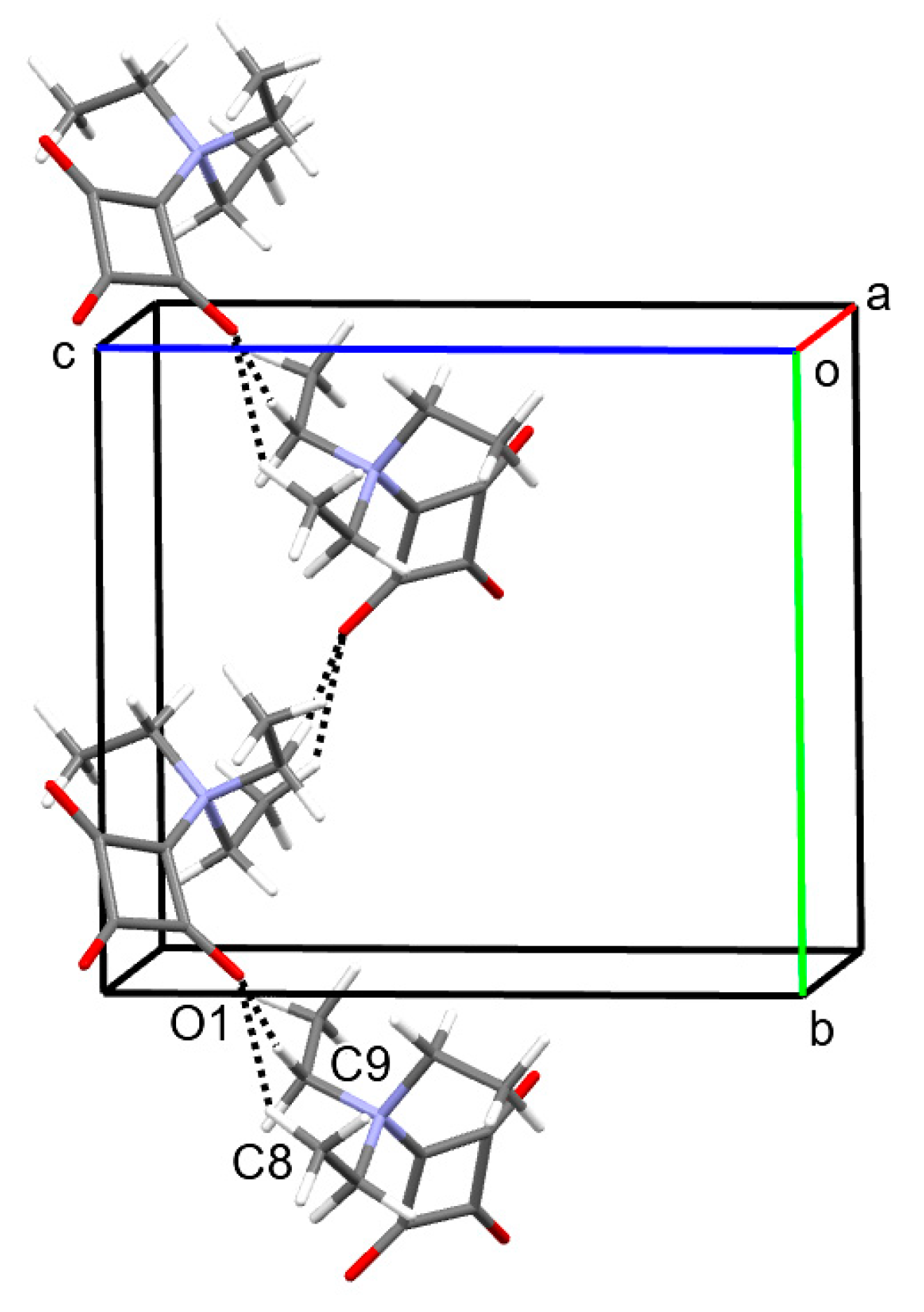
2.1. Structural Description of 1 in the Solid-State
2.2. FT-IR Spectroscopy
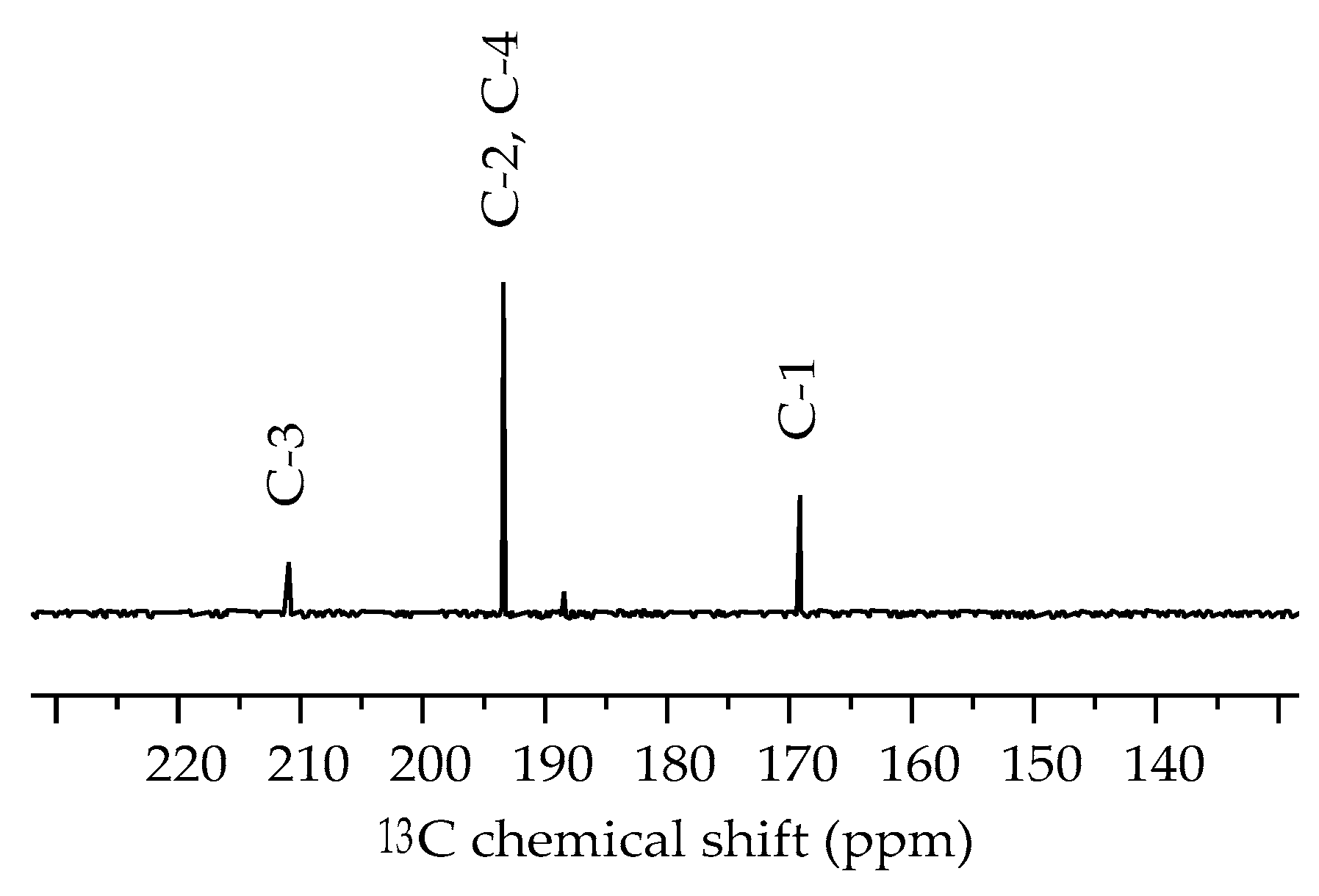
2.3. NMR Spectroscopy
3. Materials and Methods
3.1. General
3.2. Synthesis and Crystallization of 1
3.3. X-ray Crystallography
3.4. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- IUPAC. Compendium of Chemical Terminology, 2nd ed.; The “Gold Book”. Compiled by A. D. McNaught and A. Wilkinson; Blackwell Scientific Publications: Oxford, UK, 1997. [Google Scholar] [CrossRef]
- Schmidt, A.H.; Aimène, A.; Schneider, M. Eine neue Klasse von Stickstoff-Betainen der Quadratsäure. Synthesis 1984, 1984, 436–439. [Google Scholar] [CrossRef]
- Schmidt, A.H.; Aimène, A. Cyclobutendione mit Triorganyl-phosphonio-Substituenten: Neue Quadratsäureabkömmlinge sowie Vertreter “push-pull” substitutierter Pseudooxokohlenstoffe. Chem. Ztg. 1983, 107, 299–304. [Google Scholar]
- Schmidt, A.H.; Botzet, D.; Straus, M. Oxokohlenstoffe und verwandte Verbindungen, XI. Eine Variante der Dreikomponenten-Reaktion zur Darstellung von Betainen der Quadratsäure. Chem. Ztg. 1986, 110, 273–275. [Google Scholar]
- Kolev, T.M.; Yancheva, D.Y.; Stoyanov, S.I. Synthesis and Spectral and Structural Elucidation of Some Pyridinium Betaines of Squaric Acid: Potential Materials for Nonlinear Optical Applications. Adv. Funct. Mater. 2004, 14, 799–805. [Google Scholar] [CrossRef]
- Kolev, T.; Stamboliyska, B.; Yancheva, D. Spectral and structural study of two acceptor-substituted pyridinium-betaines of squaric acid: Promising chromophores for nonlinear optical applications. Chem. Phys. 2006, 324, 489–496. [Google Scholar] [CrossRef]
- Kolev, T.M.; Yancheva, D.Y.; Stamboliyska, B.A.; Dimitrov, M.D.; Wortmann, R. Nonlinear optical properties of pyridinium-betaines of squaric acid: Experimental and theoretical study. Chem. Phys. 2008, 348, 45–52. [Google Scholar] [CrossRef]
- Courbon, G.M.; Palme, P.R.; Mann, L.; Richter, A.; Imming, P.; Rubinstein, J.L. Mechanism of mycobacterial ATP synthase inhibition by squaramides and second generation diarylquinolines. EMBO J. 2023, 42, e113687. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Ranganathan, A.; Kulkarni, G.U. An Experimental Electron Density Investigation of Squarate and Croconate Dianions. J. Phys. Chem. A 2002, 106, 7813–7819. [Google Scholar] [CrossRef]
- Kolev, T.; Yancheva, D.; Schürmann, M.; Kleb, D.-C.; Preut, H.; Spiteller, M. 4-Dimethylaminopyridinium-1-squarate. Acta Crystallogr. E 2002, 58, o1267–o1268. [Google Scholar] [CrossRef]
- Kolev, T.; Yancheva, D.; Shivachev, B.; Petrova, R.; Spiteller, M. 2-(3-Benzoyl-1-pyridinio)-3,4-dioxocyclobutenolate. Acta Crystallogr. C 2005, 61, o213–o215. [Google Scholar] [CrossRef] [PubMed]
- Kolev, T.; Wortmann, R.; Spiteller, M.; Sheldrick, W.S.; Mayer-Figge, H. 4-Methoxypyridinium betaine of squaric acid. Acta Crystallogr. E 2004, 60, o1449–o1450. [Google Scholar] [CrossRef]
- Grünefeld, J.; Kunick, C.; Jones, P.G. 1-(Imidazo[1,2-a]pyridin-1-ium-1-yl)-2,3,4-trioxocyclobutan-1-ide. Molbank 2019, 2019, M1072. [Google Scholar] [CrossRef]
- Kolev, T.; Yancheva, D.; Kleb, D.C.; Preut, H.; Bleckmann, P. Crystal structure of 4-benzoylpyridinium-1-squarate, C16H9NO4. Z. Kristallogr. NCS 2001, 216, 65–66. [Google Scholar] [CrossRef]
- Kolev, T.; Yancheva, D.; Shivachev, B.; Petrova, R. The pyridinium-betaine of squaric acid. Acta Crystallogr. E 2007, 63, o3259. [Google Scholar] [CrossRef]
- Kolev, T.; Wortmann, R.; Spiteller, M.; Sheldrick, W.S.; Mayer-Figge, H. 4-Phenylpyridinium betaine of squaric acid. Acta Crystallogr. E 2005, 61, o1090–o1092. [Google Scholar] [CrossRef]
- Bestmann, H.J.; Fürst, T.G.; Schier, A. Structures and Reactions of the Oxidation Products of Dimeric Ketenylidene(triphenyl)phosphorane. Angew. Chem. Int. Ed. 1993, 32, 1746–1747. [Google Scholar] [CrossRef]
- Ucar, I.; Bulut, A.; Yesilel, O.Z.; Odabasoglu, M.; Buyukgungor, O. 3-Acetoxy-2-(acetylamino)pyridinium-1-squarate. Acta Crystallogr. C 2005, 61, o148–o150. [Google Scholar] [CrossRef]
- Sanna, E.; Martínez, L.; Rotger, C.; Blasco, S.; González, J.; García-España, E.; Costa, A. Squaramide-Based Reagent for Selective Chromogenic Sensing of Cu(II) through a Zwitterion Radical. Org. Lett. 2010, 12, 3840–3843. [Google Scholar] [CrossRef]
- Portell, A.; Font-Bardia, M.; Prohens, R. Self-Assembling of Zwitterionic Squaramides through Electrostatically Compressed Face-to-Face π-Stacking: A New Supramolecular Synthon. Cryst. Growth Des. 2013, 13, 4200–4203. [Google Scholar] [CrossRef]
- Reis, F.D.D.; Gatti, I.C.; Garcia, H.C.; de Oliveira, V.E.; de Oliveira, L.F.C. Squaraines: Crystal Structures and Spectroscopic Analysis of Hydrated and Anhydrous Forms of Squaric Acid-Isoniazid Species. J. Phys. Chem. A 2014, 118, 11521–11528. [Google Scholar] [CrossRef]
- Prohens, R.; Portell, A.; Font-Bardia, M.; Bauzá, A.; Frontera, A. A combined crystallographic and theoretical study of weak intermolecular interactions in crystalline squaric acid esters and amides. CrystEngComm 2017, 19, 3071–3077. [Google Scholar] [CrossRef]
- Kitajgorodskij, A.I. Molecular Crystals and Molecules; Academic Press: New York, NY, USA, 1973. [Google Scholar]
- Thakuria, R.; Sarma, B.; Nangia, A. 7.03—Hydrogen Bonding in Molecular Crystals. In Comprehensive Supramolecular Chemistry II; Atwood, J.L., Ed.; Elsevier: Oxford, UK, 2017; pp. 25–48. [Google Scholar]
- Kolev, T.M.; Stamboliyska, B.A.; Yancheva, D.Y.; Enchev, V. Experimental and computational studies of the structure and vibrational spectra of 4-dimethylamino pyridinium-betaine of squaric acid. J. Mol. Struct. 2004, 691, 241–248. [Google Scholar] [CrossRef]
- Kolev, T.M.; Yancheva, D.Y.; Stamboliyska, B.A. Experimental and computational studies of the structure and vibrational spectra of pyridinium-betaine of squaric acid. Spectrochim. Acta A 2003, 59, 1805–1813. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.P.; Radom, L. Harmonic Vibrational Frequencies: An Evaluation of Hartree−Fock, Møller−Plesset, Quadratic Configuration Interaction, Density Functional Theory, and Semiempirical Scale Factors. J. Phys. Chem. 1996, 100, 16502–16513. [Google Scholar] [CrossRef]
- Gurst, J.E.; Schubert, E.M.; Boiadjiev, S.E.; Lightner, D.A. Transannular Orbital Interaction in Diketones Detected by C-13 NMR Spectroscopy. Tetrahedron 1993, 49, 9191–9196. [Google Scholar] [CrossRef]
- APEX, 4.1.0; Bruker AXS Inc.: Madison, WI, USA, 2017.
- SAINT, V8.40B; Bruker AXS Inc.: Madison, WI, USA, 2019.
- SADABS, 2016/2; Bruker AXS Inc.: Madison, WI, USA, 2012.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Kleemiss, F.; Dolomanov, O.V.; Bodensteiner, M.; Peyerimhoff, N.; Midgley, L.; Bourhis, L.J.; Genoni, A.; Malaspina, L.A.; Jayatilaka, D.; Spencer, J.L.; et al. Accurate crystal structures and chemical properties from NoSpherA2. Chem. Sci. 2021, 12, 1675–1692. [Google Scholar] [CrossRef]
- Midgley, L.; Bourhis, L.J.; Dolomanov, O.V.; Grabowsky, S.; Kleemiss, F.; Puschmann, H.; Peyerimhoff, N. Vanishing of the atomic form factor derivatives in non-spherical structural refinement—A key approximation scrutinized in the case of Hirshfeld atom refinement. Acta Crystallogr. A 2021, 77, 519–533. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment—Olex2 dissected. Acta Crystallogr. A 2015, 71, 59–75. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Jayatilaka, D.; Grimwood, D.J. Tonto: A Fortran Based Object-Oriented System for Quantum Chemistry and Crystallography. In Lecture Notes in Computer Science, Computational Science—ICCS 2003; Sloot, P.M.A., Abramson, D., Bogdanov, A.V., Gorbachev, Y.E., Dongarra, J.J., Zomaya, A.Y., Eds.; Springer: Berlin/Heidelberg, Germany, 2003; pp. 142–151. [Google Scholar]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Hertwig, R.H.; Koch, W. On the parameterization of the local correlation functional. What is Becke-3-LYP? Chem. Phys. Lett. 1997, 268, 345–351. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Fletcher, R. Practical Methods of Optimization, 2nd ed.; John Wiley & Sons: Chichester, UK, 1987. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| X-ray | DFT | |
|---|---|---|
| C1−C2 | 1.4351(4) | 1.430 |
| C1−C4 | 1.4364(4) | 1.435 |
| C2−C3 | 1.5383(4) | 1.559 |
| C3−C4 | 1.5380(5) | 1.566 |
| C1−N1 | 1.4521(4) | 1.461 |
| C2−O1 | 1.2236(4) | 1.220 |
| C3−O2 | 1.2035(4) | 1.193 |
| C4−O3 | 1.2249(4) | 1.217 |
| C4−C1−C2 | 96.14(3) | 97.80 |
| N1−C1−C2 | 130.27(3) | 127.68 |
| N1−C1−C4 | 133.30(3) | 134.33 |
| C3−C2−C1 | 87.93(2) | 87.58 |
| O1−C2−C1 | 137.52(3) | 135.51 |
| O1−C2−C3 | 134.49(3) | 136.90 |
| C4−C3−C2 | 87.96(2) | 87.45 |
| O2−C3−C2 | 136.12(3) | 136.59 |
| O2−C3−C4 | 135.82(3) | 136.94 |
| C3−C4−C1 | 87.89(2) | 87.14 |
| O3−C4−C1 | 138.00(3) | 137.18 |
| O3−C4−C3 | 134.06(3) | 135.67 |
| D−H···A 1 | d(D−H) | d(H···A) | d(D···A) | <(DHA) |
|---|---|---|---|---|
| C8−H8c···O1i | 1.080(4) | 2.368(5) | 3.2624(4) | 139.1(4) |
| C9−H9b···O1i | 1.080(4) | 2.328(4) | 3.3996(4) | 171.2(4) |
| ATR | DFT 1 | Assignment 2 | Symmetry |
|---|---|---|---|
| 1789 | 1778 | C3−O2 stretch | A1 |
| 1735 | 1750 | C2−O1 and C4−O3 symmetric stretch | A1 |
| 1614 | 1633 | C2−O1 and C4−O3 asymmetric stretch | B2 |
| δ (Methanol-d4) | GIAO 1 | Assignment 2 |
|---|---|---|
| 211.0 | 222.7 | C3 |
| 193.4 | 200.9, 196.9 | C2, C4 |
| 169.1 | 174.5 | C1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palme, P.R.; Goddard, R.; Leutzsch, M.; Richter, A.; Imming, P.; Seidel, R.W. Structural Elucidation of the Triethylammonium Betaine of Squaric Acid. Molbank 2023, 2023, M1737. https://doi.org/10.3390/M1737
Palme PR, Goddard R, Leutzsch M, Richter A, Imming P, Seidel RW. Structural Elucidation of the Triethylammonium Betaine of Squaric Acid. Molbank. 2023; 2023(4):M1737. https://doi.org/10.3390/M1737
Chicago/Turabian StylePalme, Paul R., Richard Goddard, Markus Leutzsch, Adrian Richter, Peter Imming, and Rüdiger W. Seidel. 2023. "Structural Elucidation of the Triethylammonium Betaine of Squaric Acid" Molbank 2023, no. 4: M1737. https://doi.org/10.3390/M1737
APA StylePalme, P. R., Goddard, R., Leutzsch, M., Richter, A., Imming, P., & Seidel, R. W. (2023). Structural Elucidation of the Triethylammonium Betaine of Squaric Acid. Molbank, 2023(4), M1737. https://doi.org/10.3390/M1737