
Mono- and Dinuclear Carbonyl Dithiolene Complexes Related to the [FeFe]-Hydrogenases


Abstract
:1. Introduction
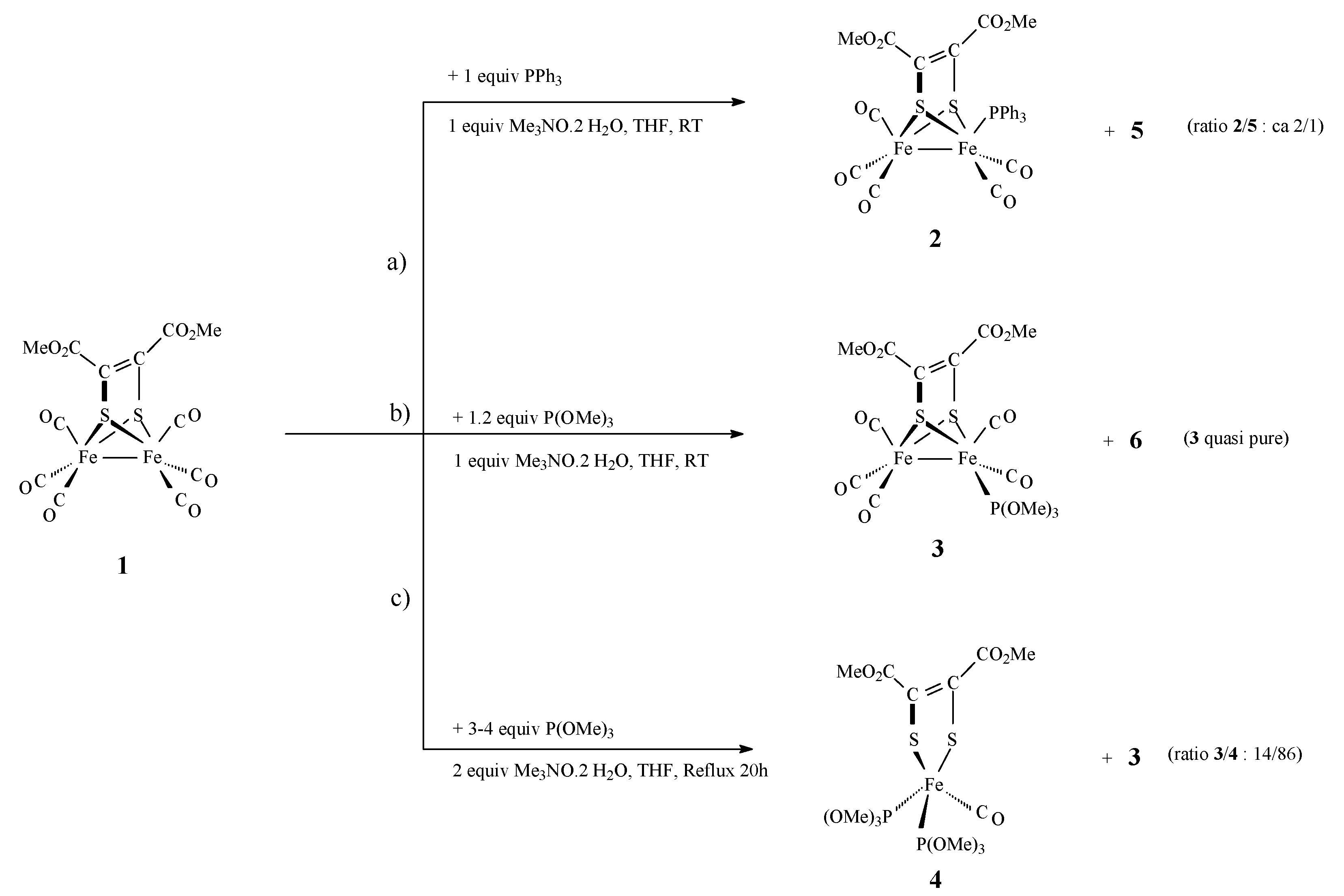
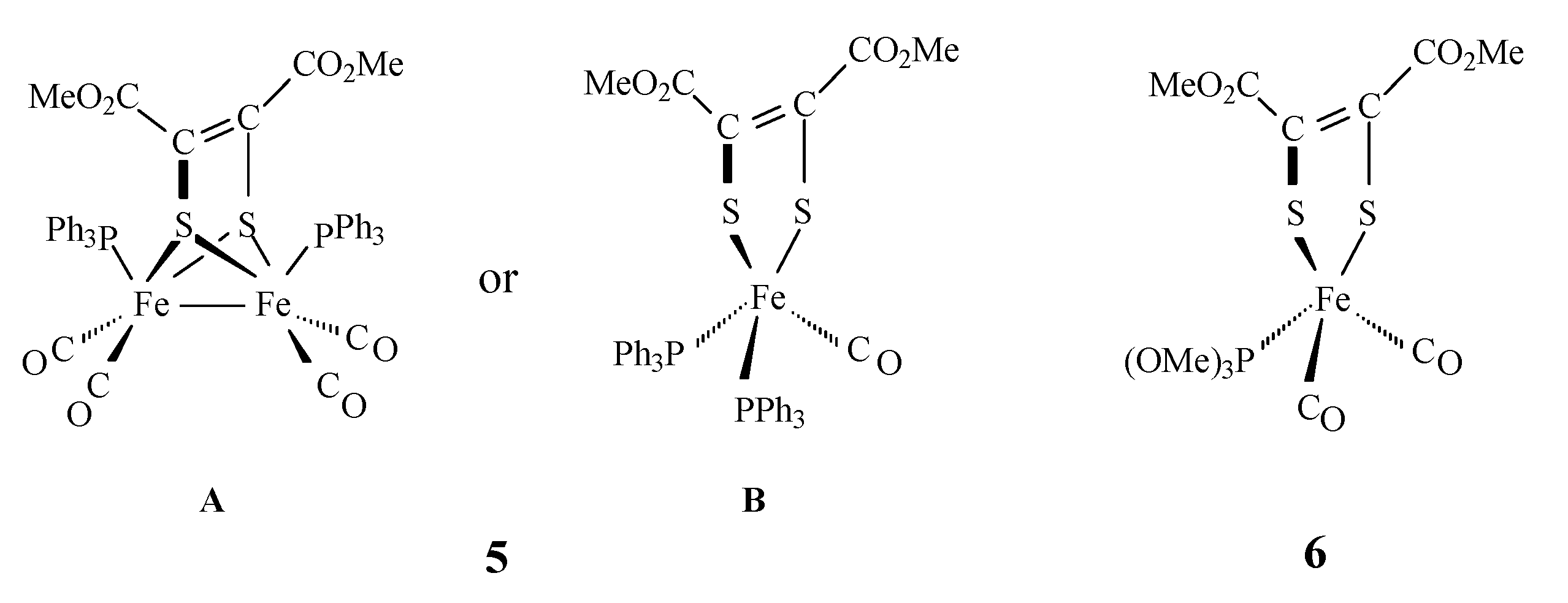
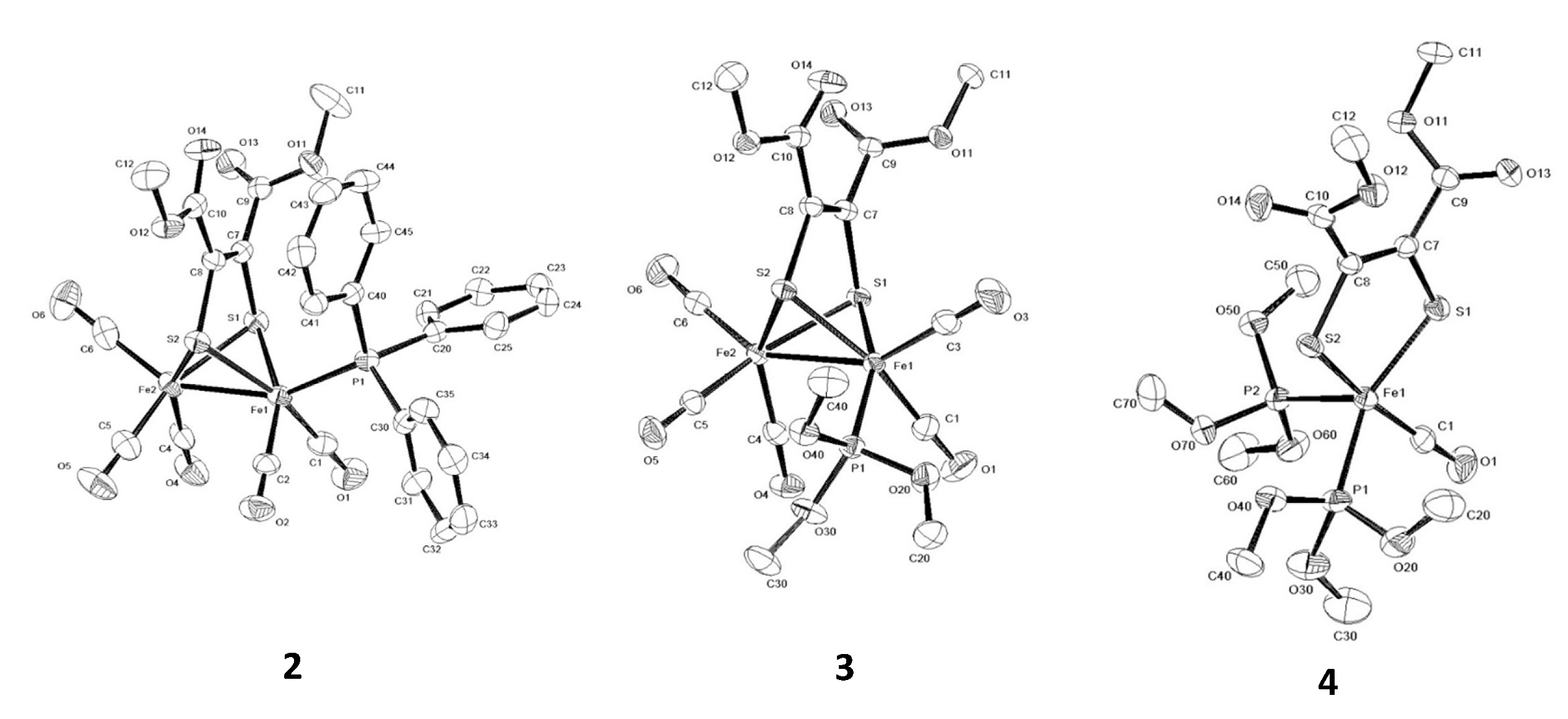
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Hogarth, G. An unexpected leading role for [Fe2(CO)6(μ-pdt)] in our understanding of [FeFe]-H2ases and the search for clean hydrogen production. Coord. Chem. Rev. 2023, 490, 215174. [Google Scholar] [CrossRef]
- Kleinhaus, J.T.; Wittkamp, F.; Yadav, S.; Siegmund, D.; Apfel, U.-P. [FeFe]-Hydrogenases: Maturation and reactivity of enzymatic systems and overview of biomimetic models. Chem. Soc. Rev. 2021, 50, 1668–1784. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Rauchfuss, T.B. Synthesis of di-iron(I) dithiolato carbonyl complexes. Chem. Rev. 2016, 116, 7043–7077. [Google Scholar] [CrossRef] [PubMed]
- Apfel, U.-P.; Pétillon, F.Y.; Schollhammer, P.; Talarmin, J.; Weigand, W. Chapter 4 [FeFe] Hydrogenase Models an Overview. In Bioinspired Catalysis; Weigand, W., Schollhammer, P., Eds.; Wiley-VCH: Weinheim, Germany, 2015; pp. 79–103. ISBN 978-3-527-33308-0. [Google Scholar]
- Elleouet, C.; Pétillon, F.Y.; Schollhammer, P. Chapter 17 [FeFe]-Hydrogenases Models. In Advances in Bioorganometallic Chemistry; Hirao, T., Moriuchi, T., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 347–364. [Google Scholar] [CrossRef]
- Elleouet, C.; Pétillon, F.Y.; Schollhammer, P. Role of a redox-active ligand close to a dinuclear activating framework. In Modes of Cooperative Effects in Dinuclear Complexes, 70; Kalck, P., Ed.; Topics in Organometallics Chemistry; Springer International Publishing: New York, NY, USA, 2022; pp. 99–156. [Google Scholar] [CrossRef]
- Charreteur, K.; Kdider, M.; Capon, J.-F.; Gloaguen, F.; Pétillon, F.Y.; Schollhammer, P.; Talarmin, J. Effect of electron-withdrawing dithiolate bridge on the electron-transfer steps in di-iron molecules related to [2Fe]H subsite of the [FeFe]-hydrogenases. Inorg. Chem. 2010, 49, 2496–2501. [Google Scholar] [CrossRef] [PubMed]
- Seyferth, D.; Henderson, R.S. Photochemically induced insertion of acetylenes into µ-dithiobis(tricarbonyliron). J. Organomet. Chem. 1979, 182, C39–C42. [Google Scholar] [CrossRef]
- Mousser, H.; Darchen, A.; Mousser, A. Unexpected fragmentation of phenyldithiobenzoate, formation and X-ray structure of [µ,η2(S,S)-1,2-(dithio)-1,2-(diphenylethylene)] di-iron hexacarbonyl complex. J. Organomet. Chem. 2010, 695, 786–791. [Google Scholar] [CrossRef]
- Boukrina Makouf, N.; Bouzidi Mousser, H.; Darchen, A.; Mousser, A. Carbon monoxide substitutions by trimethyl phosphite in di-iron dithiolate complex: Fe-Fe bond cleavage, selectivity of the substitutions, crystal structures and electrochemical studies. J. Organomet. Chem. 2018, 866, 35–42. [Google Scholar] [CrossRef]
- Adams, H.; Morris, M.J.; Robertson, C.C.; Tunnicliffe, H.C.I. Synthesis of mono- and di-iron dithiolene complexes as hydrogenase models by dithiolene transfer reactions, including the crystal structure of [{Ni(S2C2Ph2)}6]. Organometallics 2019, 38, 665–676. [Google Scholar] [CrossRef]
- Radu, L.-F.; Attia, A.A.A.; Silaghi-Dumitrescu, R.; Lupan, A. Binuclear ethylenedithiolate iron carbonyls: A density functional theory study. Inorg. Chim. Acta 2021, 519, 120260. [Google Scholar] [CrossRef]
- Almazahreh, L.R.; Imhof, W.; Talarmin, J.; Schollhammer, P.; Görls, H.; El-khateeb, M.; Weigand, W. Ligand Effects on the electrochemical behavior of [Fe2(CO)5(L){μ-(SCH2)2(Ph)P=O}] (L = PPh3, P(OEt)3) hydrogenase model complexes. Dalton Trans. 2015, 44, 7177–7199. [Google Scholar] [CrossRef] [PubMed]
- Tolman, C.A. Steric effects of phosphorus ligands in organometallic chemistry and homogeneous catalysis. Chem. Rev. 1977, 77, 313–348. [Google Scholar] [CrossRef]
- Tolman, C.A. Phosphorus ligand exchange equilibriums on zerovalent nickel. Dominant role for steric effects. J. Am. Chem. Soc. 1970, 92, 2956–2965. [Google Scholar] [CrossRef]
- Almazahreh, L.R.; Apfel, U.-P.; Imhof, W.; Rudolph, M.; Görls, H.; Talarmin, J.; Schollhammer, P.; El-khateeb, M.; Weigand, W. A novel [FeFe] hydrogenase model with a (SCH2)2P=O moiety. Organometallics 2013, 32, 4523–4530. [Google Scholar] [CrossRef]
- Rahman, M.-M.; Liu, H.-Y.; Eriks, K.; Prock, A.; Giering, W.P. Separation of phosphorus(III) ligands into pure σ-donors and σ-donor/π-acceptors: Comparison of basicity and donicity. Organometallics 1989, 8, 1–7. [Google Scholar] [CrossRef]
- Abul-Futouh, H.; Costabel, D.; Hotzel, K.; Liebing, P.; Görls, H.; Weigand, W.; Peneva, K. Mono- and di-substituted [FeFe]-hydrogenase H-cluster mimics bearing the 3,4-dimercaptobenzaldehyde bridge moiety: Insight into synthesis, characterization and electrochemical investigations. Inorg. Chim. Acta 2023, 551, 121469. [Google Scholar] [CrossRef]
- Addison, A.W.; Rao, T.N.; Reedijks, J.; van Rijn, J.; Vershoor, G.C. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc. Dalton Trans. 1984, 7, 1349–1356. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX suite for smallmolecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2 | 3 | 4 | |
|---|---|---|---|
| Fe(1)-Fe(2) | 2.4834 (6) | 2.4772 (6) | |
| P(1)-Fe(1) | 2.2496 (8) | 2.1651 (9) | 2.1621 (9) |
| P(2)-Fe(1) | 2.1214 (9) | ||
| S(1)-Fe(1) | 2.2852 (8) | 2.2762 (9) | 2.1762 (8) |
| S(1)-Fe(2) | 2.2886 (8) | 2.2908 (9) | |
| S(2)-Fe(1) | 2.2872 (8) | 2.2810 (9) | 2.1983 (8) |
| S(2)-Fe(2) | 2.2742 (8) | 2.2667 (9) | |
| C(1)-Fe(1) | 1.771 (3) | 1.786 (4) | 1.768 (3) |
| C(2/3)-Fe(1) | 1.769 (3) | 1.778 (4) | |
| C(4)-Fe(2) | 1.794 (3) | 1.789 (3) | |
| C(5)-Fe(2) | 1.776 (3) | 1.793 (3) | |
| C(6)-Fe(2) | 1.812 (3) | 1.811 (3) | |
| C(7)-C(8) | 1.323 (4) | 1.329 (4) | 1.354 (4) |
| Fe(1)-S(1)-Fe(2) | 65.77 (2) | 65.69 (3) | |
| Fe(1)-S(2)-Fe(2) | 65.97 (2) | 66.01 (3) | |
| C(1)-Fe(1)-S(1) | 90.95 (9) | 90.42 (11) | |
| C(1)-Fe(1)-S(2) | 161.07 (10) | 163.16 (12) | 170.23 (10) |
| C(2/3)-Fe(1)-S(1) | 156.84 (10) | 105.76 (12) | |
| P(1)-Fe(1)-S(1) | 106.47 (3) | 156.95 (4) | 151.49 (4) |
| P(2)-Fe(1)-S(1) | 116.47 (4) | ||
| P(1)-Fe(1)-S(2) | 102.98 (3) | 95.07 (3) | |
| P(2)-Fe(1)-S(2) | 96.24 (3) | ||
| P(1)-Fe(1)-C(1) | 95.94 (10) | 87.60 (11) | 90.95 (11) |
| P(2)-Fe(1)-C(1) | 93.47 (10) | ||
| P(1)-Fe(1)-C(2/3) | 96.26 (9) | 97.27 (12) | |
| P(1)-Fe(1)-P(2) | 93.98 (3) | ||
| C(1)-Fe(1)-C(2/3) | 91.30 (3) | 97.09 (17) | |
| S(1)-Fe(1)-S(2) | 79.80 (9) | 80.62 (3) | |
| C(4)-Fe(2)-C(5) | 91.19 (13) | 90.11 (14) | |
| C(4)-Fe(2)-S(1) | 91.50 (9) | 92.81 (10) | |
| C(4)-Fe(2)-S(2) | 154.58 (10) | 146.25 (11) | |
| C(5)-Fe(2)-S(1) | 159.48 (11) | 164.82 (11) | |
| C(5)-Fe(2)-S(2) | 88.98 (9) | 88.71 (11) | |
| C(6)-Fe(2)-Fe(1) | 149.44 (10) | 148.77 (11) | |
| P(1)/C(3)-Fe(1)-Fe(2) | 153.93 (3) | 150.45 (12) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kdider, M.; Elleouet, C.; Pétillon, F.Y.; Schollhammer, P. Mono- and Dinuclear Carbonyl Dithiolene Complexes Related to the [FeFe]-Hydrogenases. Molbank 2023, 2023, M1719. https://doi.org/10.3390/M1719
Kdider M, Elleouet C, Pétillon FY, Schollhammer P. Mono- and Dinuclear Carbonyl Dithiolene Complexes Related to the [FeFe]-Hydrogenases. Molbank. 2023; 2023(3):M1719. https://doi.org/10.3390/M1719
Chicago/Turabian StyleKdider, Mohamed, Catherine Elleouet, François Y. Pétillon, and Philippe Schollhammer. 2023. "Mono- and Dinuclear Carbonyl Dithiolene Complexes Related to the [FeFe]-Hydrogenases" Molbank 2023, no. 3: M1719. https://doi.org/10.3390/M1719
APA StyleKdider, M., Elleouet, C., Pétillon, F. Y., & Schollhammer, P. (2023). Mono- and Dinuclear Carbonyl Dithiolene Complexes Related to the [FeFe]-Hydrogenases. Molbank, 2023(3), M1719. https://doi.org/10.3390/M1719