
Tributyl(1-((dimethylamino)(dimethyliminio)methyl)-1,4-dihydropyridin-4-yl)phosphonium Ditrifluoromethanesulfonate



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
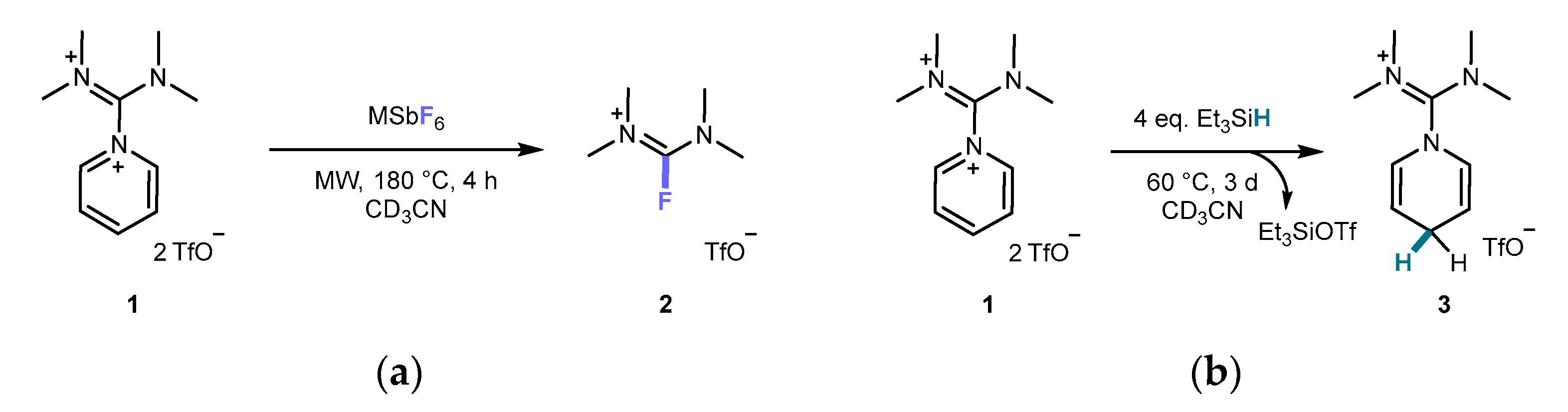
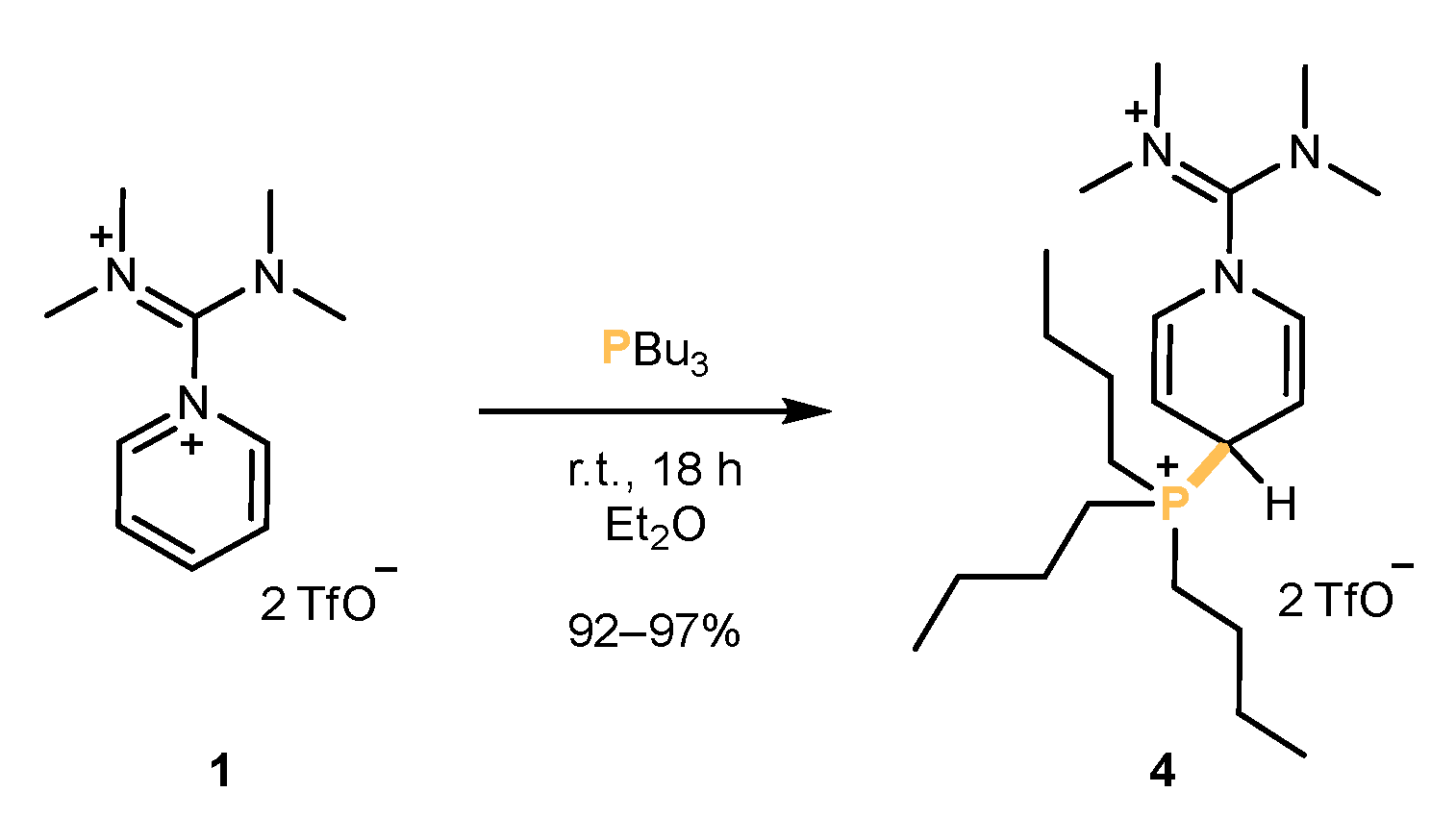
2.1. Synthesis
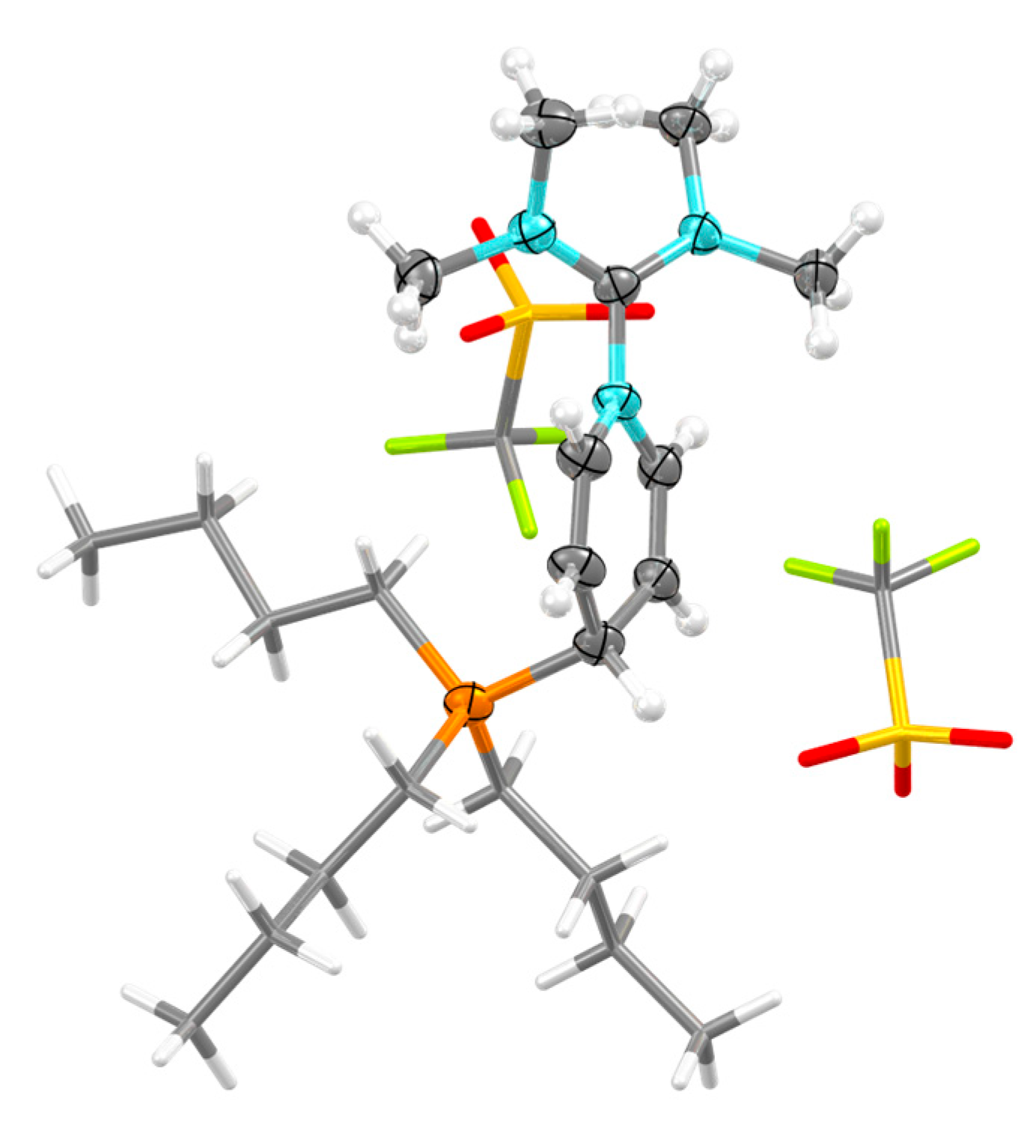
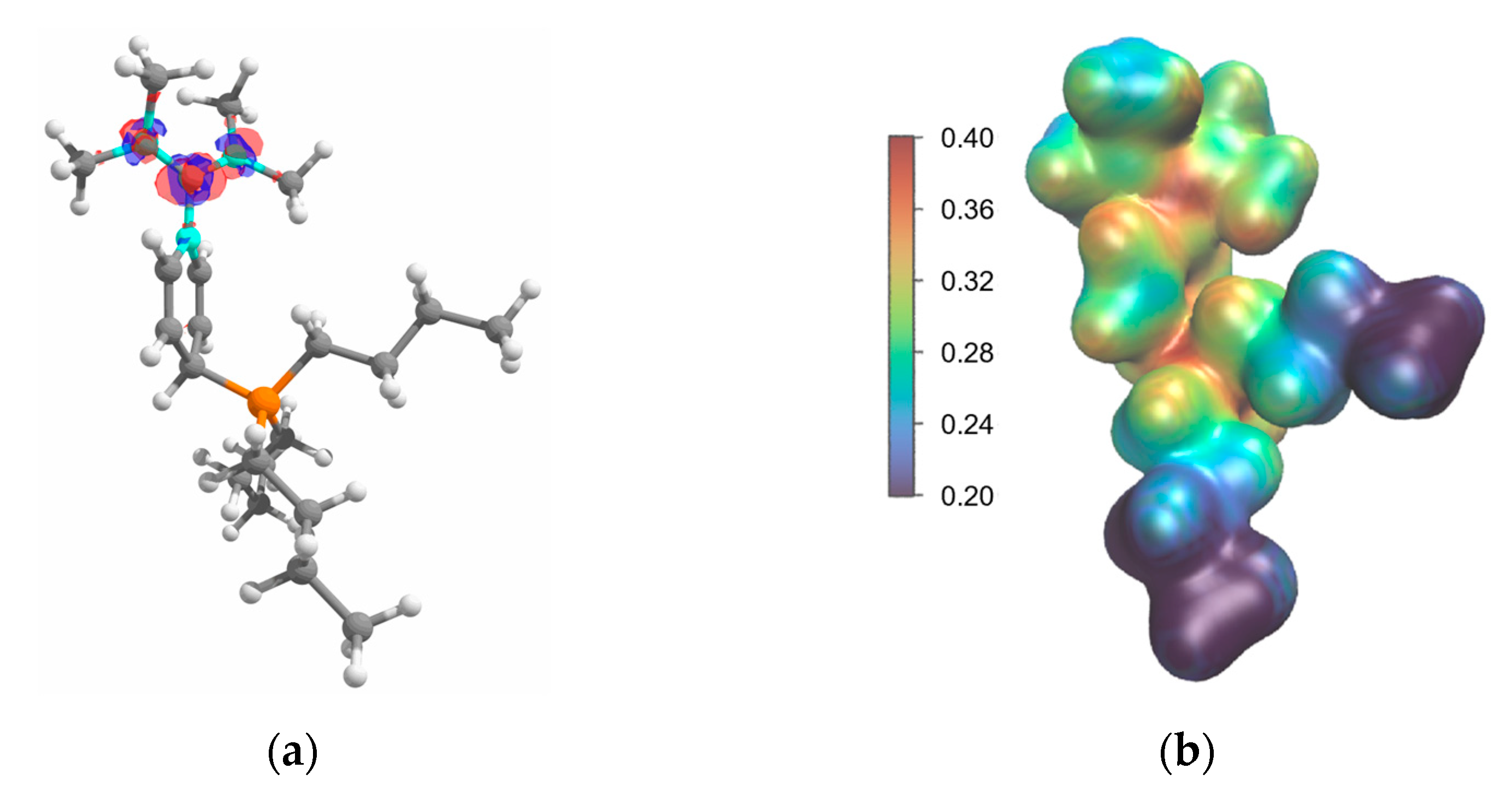
2.2. Structure in the Solid State
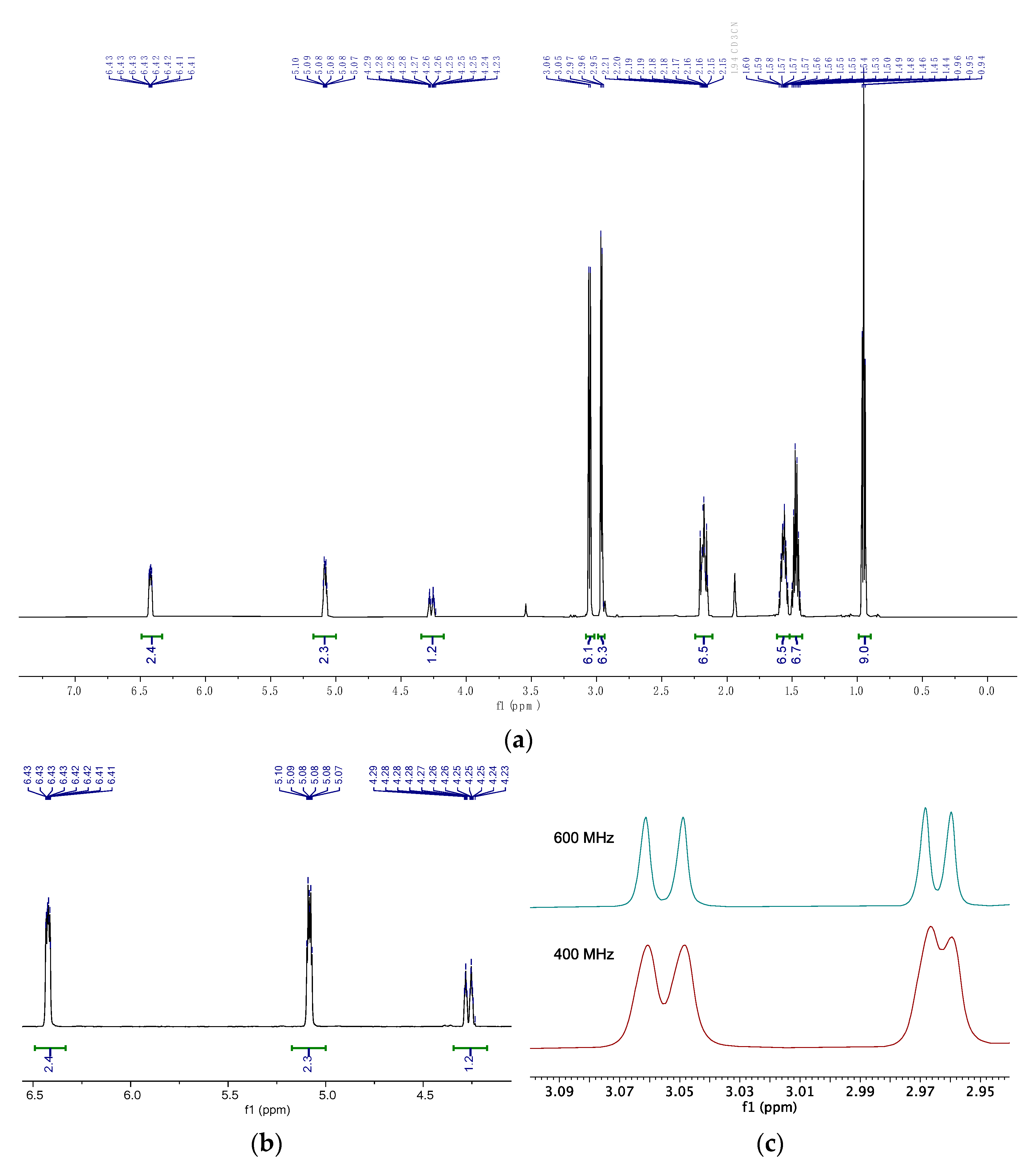
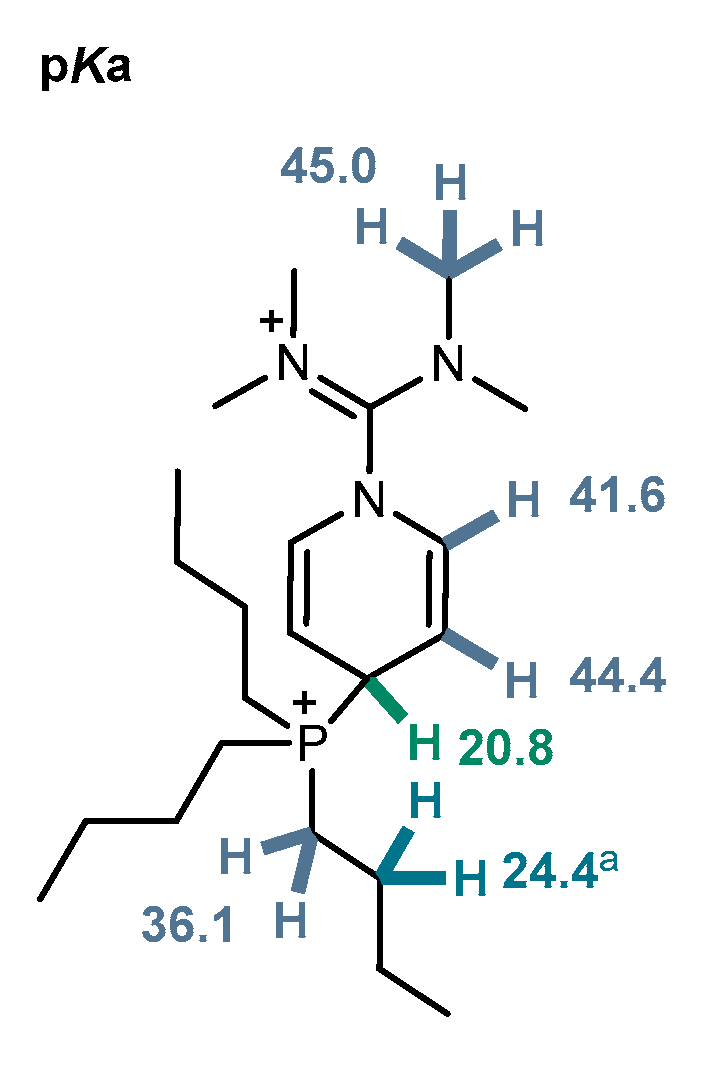
2.3. Nuclear Magnetic Resonance
2.4. Computational Investigation
3. Materials and Methods
3.1. Experimental Methods
3.2. Computational Methods
3.3. Synthesis of 4
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef]
- Stephens, D.E.; Larionov, O.V. Recent Advances in the C-H-Functionalization of the Distal Positions in Pyridines and Quinolines. Tetrahedron 2015, 71, 8683–8716. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Zhou, F.-Y. Recent Developments in Transition-Metal-Free Functionalization and Derivatization Reactions of Pyridines. Synlett 2020, 32, 159–178. [Google Scholar] [CrossRef]
- Maity, S.; Bera, A.; Bhattacharjya, A.; Maity, P. C-H functionalization of pyridines. Org. Biomol. Chem. 2023, 21, 5671–5690. [Google Scholar] [CrossRef]
- Kim, M.; Koo, Y.; Hong, S. N-Functionalized Pyridinium Salts: A New Chapter for Site-Selective Pyridine C-H Functionalization via Radical-Based Processes under Visible Light Irradiation. Acc. Chem. Res. 2022, 55, 3043–3056. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Mathi, G.R.; Hong, S.; Hong, S. Enantioselective functionalization at the C4 position of pyridinium salts through NHC catalysis. Nat. Commun. 2022, 13, 1776. [Google Scholar] [CrossRef]
- Ma, X.; Herzon, S.B. Intermolecular Hydropyridylation of Unactivated Alkenes. J. Am. Chem. Soc. 2016, 138, 8718–8721. [Google Scholar] [CrossRef]
- Fier, P.S. A Bifunctional Reagent Designed for the Mild, Nucleophilic Functionalization of Pyridines. J. Am. Chem. Soc. 2017, 139, 9499–9502. [Google Scholar] [CrossRef]
- Mathi, G.R.; Kweon, B.; Moon, Y.; Jeong, Y.; Hong, S. Regioselective C−H Functionalization of Heteroarene N-Oxides Enabled by a Traceless Nucleophile. Angew. Chem. Int. Ed. 2020, 59, 22675–22683. [Google Scholar] [CrossRef]
- Choi, J.; Laudadio, G.; Godineau, E.; Baran, P.S. Practical and Regioselective Synthesis of C-4-Alkylated Pyridines. J. Am. Chem. Soc. 2021, 143, 11927–11933. [Google Scholar] [CrossRef] [PubMed]
- Bull, J.A.; Mousseau, J.J.; Pelletier, G.; Charette, A.B. Synthesis of Pyridine and Dihydropyridine Derivatives by Regio- and Stereoselective Addition to N-Activated Pyridines. Chem. Rev. 2012, 112, 2642–2713. [Google Scholar] [CrossRef]
- Liu, Z.; He, J.-H.; Zhang, M.; Shi, Z.-J.; Tang, H.; Zhou, X.-Y.; Tian, J.-J.; Wang, X.-C. Borane-Catalyzed C3-Alkylation of Pyridines with Imines, Aldehydes, or Ketones as Electrophiles. J. Am. Chem. Soc. 2022, 144, 4810–4818. [Google Scholar] [CrossRef]
- Muta, R.; Torigoe, T.; Kuninobu, Y. 3-Position-Selective C–H Trifluoromethylation of Pyridine Rings Based on Nucleophilic Activation. Org. Lett. 2022, 24, 8218–8222. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-Y.; Zhang, M.; Liu, Z.; He, J.-H.; Wang, X.-C. C3-Selective Trifluoromethylthiolation and Difluoromethylthiolation of Pyridines and Pyridine Drugs via Dihydropyridine Intermediates. J. Am. Chem. Soc. 2022, 144, 14463–14470. [Google Scholar] [CrossRef]
- Liu, Z.; Shi, Z.-J.; Liu, L.; Zhang, M.; Zhang, M.-C.; Guo, H.-Y.; Wang, X.-C. Asymmetric C3-Allylation of Pyridines. J. Am. Chem. Soc. 2023, 145, 11789–11797. [Google Scholar] [CrossRef] [PubMed]
- Birch, A.J.; Karakhanov, E.A. Preparation of some N-substituted 1,4-dihydropyridines by metal–ammonia reactions. J. Chem. Soc. Chem. Commun. 1975, 12, 480–481. [Google Scholar] [CrossRef]
- Schlosser, M.; Schneider, P. Metalation of Pyrans and Dihydropyridines: When is an 8 pi-System Cost Effective? Angew. Chem. Int. Ed. 1979, 18, 489–490. [Google Scholar] [CrossRef]
- De Koning, A.J.; Budzelaar, P.H.M.; Brandsma, L.; de Bie, M.J.A.; Boersma, J. Synthesis and NMR spectroscopic properties of some methyl-substituted 1-methyl-1,4-dihydropyridines. Tetrahedron Lett. 1980, 21, 2105–2108. [Google Scholar] [CrossRef]
- Akiyama, K.; Ishii, T.; Tero-Kubota, S.; Ikegami, Y. Photolytic Generation and the Subsequent Dimerization of 4-Alkyl-1-methylpyridinyl Radicals in Solution as Studied by Steady-state and Kinetic ESR Spectroscopy. Bull. Chem. Soc. Jpn. 1985, 58, 3535–3539. [Google Scholar] [CrossRef]
- Stephan, D.W. Frustrated Lewis Pairs. J. Am. Chem. Soc. 2015, 137, 10018–10032. [Google Scholar] [CrossRef]
- Jupp, A.R.; Stephan, D.W. New Directions for Frustrated Lewis Pair Chemistry. Trends Chem. 2019, 1, 35–48. [Google Scholar] [CrossRef]
- Mahdi, T.; del Castillo, J.N.; Stephan, D.W. Metal-Free Hydrogenation of N-Based Heterocycles. Organometallics 2013, 32, 1971–1978. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhang, L.; Meng, W.; Feng, X.; Yang, J.; Du, H. Borane-Catalyzed Transfer Hydrogenations of Pyridines with Ammonia Borane. Org. Lett. 2016, 18, 5189–5191. [Google Scholar] [CrossRef]
- Eisenberger, P.; Bestvater, B.P.; Keske, E.C.; Crudden, C.M. Hydrogenations at Room Temperature and Atmospheric Pressure with Mesoionic Carbene-Stabilized Borenium Catalysts. Angew. Chem. Int. Ed. 2015, 54, 2467–2471. [Google Scholar] [CrossRef]
- Yang, Z.-Y.; Luo, H.; Zhang, M.; Wang, X.-C. Borane-Catalyzed Reduction of Pyridines via a Hydroboration/Hydrogenation Cascade. ACS Catal. 2021, 11, 10824–10829. [Google Scholar] [CrossRef]
- Fan, X.; Zheng, J.; Li, Z.H.; Wang, H. Organoborane Catalyzed Regioselective 1,4-Hydroboration of Pyridines. J. Am. Chem. Soc. 2015, 137, 4916–4919. [Google Scholar] [CrossRef]
- Gandhamsetty, N.; Park, S.; Chang, S. Selective Silylative Reduction of Pyridines Leading to Structurally Diverse Azacyclic Compounds with the Formation of sp3 C–Si Bonds. J. Am. Chem. Soc. 2015, 137, 15176–15184. [Google Scholar] [CrossRef]
- Liu, Z.-Y.; Wen, Z.-H.; Wang, X.-C. B(C6F5)3-Catalyzed Cascade Reduction of Pyridines. Angew. Chem. Int. Ed. 2017, 56, 5817–5820. [Google Scholar] [CrossRef]
- Greßies, S.; Süße, L.; Casselman, T.; Stoltz, B.M. Tandem Dearomatization/Enantioselective Allylic Alkylation of Pyridines. J. Am. Chem. Soc. 2023, 145, 11907–11913. [Google Scholar] [CrossRef]
- Bormann, N.; Ward, J.S.; Bergmann, A.K.; Wenz, P.; Rissanen, K.; Gong, Y.; Hatz, W.-B.; Burbaum, A.; Mulks, F.F. Diiminium Nucleophile Adducts are Stable and Convenient Strong Lewis Acids. Chem. Eur. J. 2023, e202302089. [Google Scholar] [CrossRef]
- Greb, L. Lewis Superacids: Classifications, Candidates, and Applications. Chem. Eur. J. 2018, 24, 17881–17896. [Google Scholar] [CrossRef]
- Anders, E.; Opitz, A.; Wermann, K.; Wiedel, B.; Walther, M.; Imhof, W.; Gorls, H. Preparation and Conversion of N-Halomethylpyridinium Halides. Comparison with Related Compounds. J. Org. Chem. 1999, 64, 3113–3121. [Google Scholar] [CrossRef]
- Ho Lee, P.; Lee, K.; Hwan Shim, J.; Guk Lee, S.; Kim, S. Regioselective Synthesis of 4-Alkylpyridines from Pyridine and Aldehydes via Dipole Reversal Process of 1,4-Dihydropyridine Phosphonate. Heterocycles 2006, 67, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Kozuch, S.; Gruzman, D.; Martin, J.M.L. DSD-BLYP: A General Purpose Double Hybrid Density Functional Including Spin Component Scaling and Dispersion Correction. J. Phys. Chem. C 2010, 114, 20801–20808. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057. [Google Scholar] [CrossRef] [PubMed]
- Hellweg, A.; Hättig, C.; Höfener, S. Optimized accurate auxiliary basis sets for RI-MP2 and RI-CC2 calculations for the atoms Rb to Rn. Theor. Chem. Acc. 2007, 117, 587–597. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Chem. Phys. 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Grimme, S.; Brandenburg, J.G.; Bannwarth, C.; Hansen, A. Consistent structures and interactions by density functional theory with small atomic orbital basis sets. J. Chem. Phys. 2015, 143, 054107. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Chem. Phys. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Kruse, H.; Grimme, S. A geometrical correction for the inter- and intra-molecular basis set superposition error in Hartree-Fock and density functional theory calculations for large systems. J. Chem. Phys. 2012, 136, 154101. [Google Scholar] [CrossRef] [PubMed]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree–Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree–Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Böhrer, H.; Trapp, N.; Himmel, D.; Schleep, M.; Krossing, I. From unsuccessful H2-activation with FLPs containing B(Ohfip) 3 to a systematic evaluation of the Lewis acidity of 33 Lewis acids based on fluoride, chloride, hydride and methyl ion affinities. Dalton Trans. 2015, 44, 7489–7499. [Google Scholar] [CrossRef]
- Erdmann, P.; Leitner, J.; Schwarz, J.; Greb, L. An Extensive Set of Accurate Fluoride Ion Affinities for p-Block Element Lewis Acids and Basic Design Principles for Strong Fluoride Ion Acceptors. ChemPhysChem 2020, 21, 987–994. [Google Scholar] [CrossRef]
- Erdmann, P.; Greb, L. Multidimensional Lewis Acidity: A Consistent Data Set of Chloride, Hydride, Methide, Water and Ammonia Affinities for 183 p-Block Element Lewis Acids. ChemPhysChem 2021, 22, 935–943. [Google Scholar] [CrossRef]
- Jupp, A.R.; Johnstone, T.C.; Stephan, D.W. The global electrophilicity index as a metric for Lewis acidity. Dalton Trans. 2018, 47, 7029–7035. [Google Scholar] [CrossRef] [PubMed]
- Christe, K.O.; Dixon, D.A.; McLemore, D.; Wilson, W.W.; Sheehy, J.A.; Boatz, J.A. On a quantitative scale for Lewis acidity and recent progress in polynitrogen chemistry. J. Fluorine Chem. 2000, 101, 151–153. [Google Scholar] [CrossRef]
- Vogler, M.; Süsse, L.; Lafortune, J.H.W.; Stephan, D.W.; Oestreich, M. Electrophilic Phosphonium Cations as Lewis Acid Catalysts in Diels-Alder Reactions and Nazarov Cyclizations. Organometallics 2018, 37, 3303–3313. [Google Scholar] [CrossRef]
- Li, M.L.; Yu, J.H.; Li, Y.H.; Zhu, S.F.; Zhou, Q.L. Highly enantioselective carbene insertion into N-H bonds of aliphatic amines. Science 2019, 366, 990–994. [Google Scholar] [CrossRef]
- Tshepelevitsh, S.; Kütt, A.; Lõkov, M.; Kaljurand, I.; Saame, J.; Heering, A.; Plieger, P.G.; Vianello, R.; Leito, I. On the Basicity of Organic Bases in Different Media. Eur. J. Org. Chem. 2019, 2019, 6735–6748. [Google Scholar] [CrossRef]
- Mnova; vesion 14.2.3; Mestrelab Research, S.L.: Santiago de Compostela, Spain, 2022.
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef] [PubMed]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Harris, R.K.; Becker, E.D.; Cabral De Menezes, S.M.; Goodfellow, R.; Granger, P. NMR nomenclature. Nuclear spin properties and conventions for chemical shifts (IUPAC Recommendations 2001). Pure Appl. Chem. 2001, 73, 1795–1818. [Google Scholar] [CrossRef]
- CrysAlisPro; Rigaku Oxford Diffraction, Rigaku Corporation: Oxford, UK, 2018.
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Young, T.A.; Silcock, J.J.; Sterling, A.J.; Duarte, F. autodE: Automated Calculation of Reaction Energy Profiles— Application to Organic and Organometallic Reactions. Angew. Chem. Int. Ed. 2021, 60, 4266–4274. [Google Scholar] [CrossRef]
- Riniker, S.; Landrum, G.A. Better Informed Distance Geometry: Using What We Know To Improve Conformation Generation. J. Chem. Inf. Model. 2015, 55, 2562–2574. [Google Scholar] [CrossRef]
- Bannwarth, C.; Caldeweyher, E.; Ehlert, S.; Hansen, A.; Pracht, P.; Seibert, J.; Spicher, S.; Grimme, S. Extended tight-binding quantum chemistry methods. Wiley Interdiscip. Rev. Comput. Mol. 2021, 11, e1493. [Google Scholar] [CrossRef]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef] [PubMed]
- Born, M. Volumen und Hydratationswärme der Ionen. Z. Phys. 1920, 1, 45–48. [Google Scholar] [CrossRef]
- Klopman, G. Solvations: A semi-empirical procedure for including solvation in quantum mechanical calculations of large molecules. Chem. Phys. Lett. 1967, 1, 200–202. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system—Version 5.0. Wiley Interdiscip. Rev. Comput. Mol. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Grimme, S. Supramolecular Binding Thermodynamics by Dispersion-Corrected Density Functional Theory. Chem. Eur. J. 2012, 18, 9955–9964. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, Y.; Ward, J.S.; Rissanen, K.; Mulks, F.F. Tributyl(1-((dimethylamino)(dimethyliminio)methyl)-1,4-dihydropyridin-4-yl)phosphonium Ditrifluoromethanesulfonate. Molbank 2023, 2023, M1710. https://doi.org/10.3390/M1710
Gong Y, Ward JS, Rissanen K, Mulks FF. Tributyl(1-((dimethylamino)(dimethyliminio)methyl)-1,4-dihydropyridin-4-yl)phosphonium Ditrifluoromethanesulfonate. Molbank. 2023; 2023(3):M1710. https://doi.org/10.3390/M1710
Chicago/Turabian StyleGong, Yiwei, Jas S. Ward, Kari Rissanen, and Florian F. Mulks. 2023. "Tributyl(1-((dimethylamino)(dimethyliminio)methyl)-1,4-dihydropyridin-4-yl)phosphonium Ditrifluoromethanesulfonate" Molbank 2023, no. 3: M1710. https://doi.org/10.3390/M1710
APA StyleGong, Y., Ward, J. S., Rissanen, K., & Mulks, F. F. (2023). Tributyl(1-((dimethylamino)(dimethyliminio)methyl)-1,4-dihydropyridin-4-yl)phosphonium Ditrifluoromethanesulfonate. Molbank, 2023(3), M1710. https://doi.org/10.3390/M1710