
1-(2-Chlorophenyl)-6,7-dimethoxy-3-methyl-3,4-dihydroisoquinoline





{kind=link}
Abstract
1. Introduction
2. Results and Discussion
2.1. In Silico Prediction of Activity
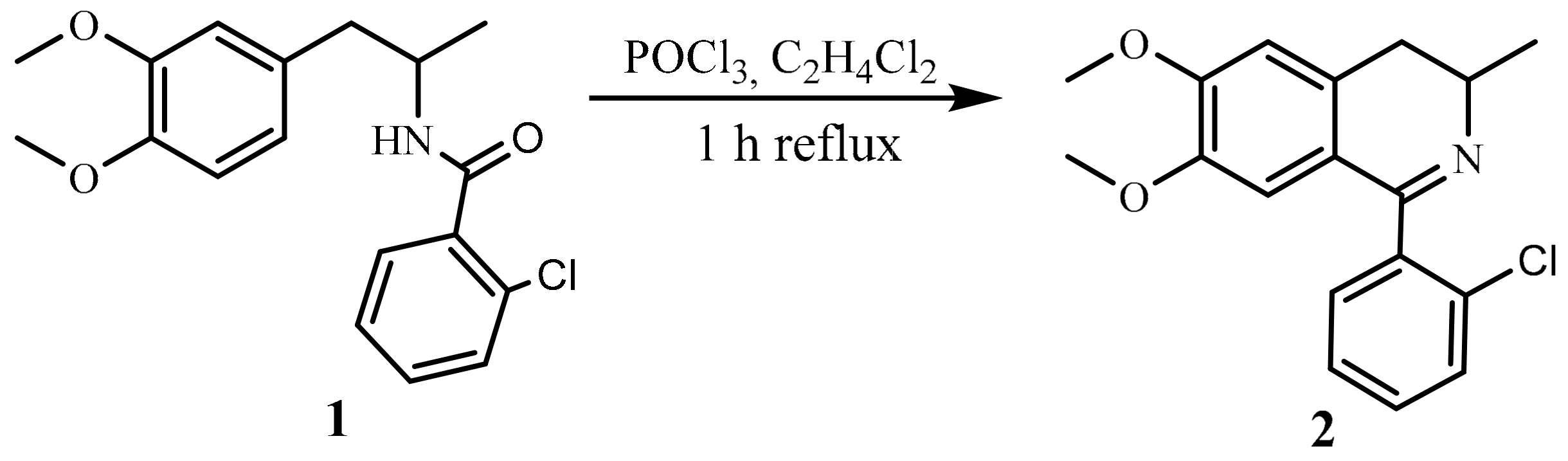
2.2. Synthesis of the Target Molecule
3. Materials and Methods
3.1. In Silico Calculations
3.1.1. Theoretical Prediction of Pharmacokinetic Parameters (ADME)
3.1.2. Theoretical Prediction of Toxicity
3.1.3. PASS Online Predictions
3.2. Synthetic Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bhadra, K.; Suresh, K.G. Therapeutic potential of nucleic acid-binding isoquinoline alkaloids: Binding aspects and implications for drug design. Med. Res. Rev. 2011, 31, 821–862. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.Y.; Kumar, G.S. Natural isoquinoline alkaloids: Binding aspects to functional proteins, serum albumins, hemoglobin, and lysozyme. Biophys. Rev. 2015, 7, 407–420. [Google Scholar] [CrossRef]
- Ivanov, I.; Nikolova, S.; Aladjov, D.; Stefanova, I.; Zagorchev, P. Synthesis and Contractile Activity of Substituted 1,2,3,4-Tetrahydroisoquinolines. Molecules 2011, 16, 7019–7042. [Google Scholar] [CrossRef]
- Kmieciak, A.; Ćwiklińska, M.; Jeżak, K.; Shili, A.; Krzemiński, M.P. Searching for New Biologically Active Compounds Derived from Isoquinoline Alkaloids. Chem. Proc. 2021, 3, 97. [Google Scholar] [CrossRef]
- Qing, Z.-X.; Yang, P.; Tang, Q.; Cheng, P.; Liu, X.-B.; Zheng, Y.J.; Liu, Y.-S.; Zeng, J.-G. Isoquinoline alkaloids and their antiviral, antibacterial, and antifungal activities and structure-activity relationship. Curr. Org. Chem. 2017, 21, 1. [Google Scholar] [CrossRef]
- Qian, J.-Q. Cardiovascular pharmacological effects of bisbenzylisoquinoline alkaloid derivatives. Acta Pharmacol. Sin. 2002, 23, 1086–1092. [Google Scholar]
- Kukula-Koch, W.; Widelski, J. Alkaloids. In Pharmacognosy; Badal, S., Delgoda, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 163–198. [Google Scholar] [CrossRef]
- Ahmed, W.; Huang, Z.-H.; Cui, Z.-N.; Tang, R.-Y. Design and synthesis of unique thiazoloisoquinolinium thiolates and derivatives. Chin. Chem. Lett. 2021, 32, 3211–3214. [Google Scholar] [CrossRef]
- Bentley, K.W. β-Phenylethylamines and the isoquinoline alkaloids. Nat. Prod. Rep. 2001, 18, 148–170. [Google Scholar] [CrossRef]
- Luo, C.; Ampomah-Wireko, M.; Wang, H.; Wu, C.; Wang, Q.; Zhang, H.; Cao, Y. Isoquinolines: Important Cores in Many Marketed and Clinical Drugs. Anticancer Agents Med. Chem. 2021, 21, 811–824. [Google Scholar] [CrossRef]
- Shaik, A.B.; Bhandare, R.R.; Nissankararao, S.; Edis, Z.; Tangirala, N.R.; Shahanaaz, S.; Rahman, M.M. Design, Facile Synthesis and Characterization of Dichloro Substituted Chalcones and Dihydropyrazole Derivatives for Their Antifungal, Antitubercular and Antiproliferative Activities. Molecules 2020, 25, 3188. [Google Scholar] [CrossRef]
- Milusheva, M.; Gledacheva, V.; Batmazyan, M.; Nikolova, S.; Stefanova, I.; Dimitrova, D.; Saracheva, K.; Tomov, D.; Chaova-Gizdakova, V. Ex Vivo and In Vivo Study of Some Isoquinoline Precursors. Sci. Pharm. 2022, 90, 37. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. Swiss ADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Martin, Y.C. A bioavailability score. J. Med. Chem. 2005, 48, 3164–3170. [Google Scholar] [CrossRef]
- Pathan, N.; Shende, P. Tailoring of P-glycoprotein for effective transportation of actives across blood-brain-barrier. J. Control. Release 2021, 335, 398–407. [Google Scholar] [CrossRef]
- Testa, B.; Kraemer, S.D. The biochemistry of drug metabolism—An introduction: Part 4. Reactions of conjugation and their enzymes. Chem. Biodivers. 2008, 5, 2171–2336. [Google Scholar] [CrossRef]
- Isyaku, Y.; Uzairu, A.; Uba, S. Computational studies of a series of 2-substituted phenyl-2-oxo-, 2-hydroxyl- and 2-acylloxyethylsulfonamides as potent anti-fungal agents. Heliyon 2020, 6, e03724. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef]
- Mazumder, K.; Hossain, E.; Aktar, A.; Mohiuddin, M.; Sarkar, K.K.; Biswas, B.; Aziz, A.; Abid, A.; Fukase, K. In Silico Analysis and Experimental Evaluation of Ester Prodrugs of Ketoprofen for Oral Delivery: With a View to Reduce Toxicity. Processes 2021, 9, 2221. [Google Scholar] [CrossRef]
- Anzali, S.; Barnickel, G.; Cezanne, B.; Krug, M.; Filimonov, D.; Poroikov, V. Discriminating between Drugs and Nondrugs by Prediction of Activity Spectra for Substances (PASS). J. Med. Chem. 2001, 44, 2432–2437. [Google Scholar] [CrossRef]
- Mathew, B.; Suresh, J.; Anbazhagan, S. Synthesis and PASS-assisted in silico approach of some novel 2-substituted ben-zim-idazole bearing a pyrimidine-2,4,6 (trione) system as mucomembranous protector. J. Pharm. Bioallied Sci 2013, 5, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Olechno, J.; Williams, A.J. Dispensing Processes Impact Apparent Biological Activity as Determined by Computational and Statistical Analyses. PLoS ONE 2013, 8, e62325. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milusheva, M.; Gledacheva, V.; Stefanova, I.; Nikolova, S. 1-(2-Chlorophenyl)-6,7-dimethoxy-3-methyl-3,4-dihydroisoquinoline. Molbank 2023, 2023, M1608. https://doi.org/10.3390/M1608
Milusheva M, Gledacheva V, Stefanova I, Nikolova S. 1-(2-Chlorophenyl)-6,7-dimethoxy-3-methyl-3,4-dihydroisoquinoline. Molbank. 2023; 2023(2):M1608. https://doi.org/10.3390/M1608
Chicago/Turabian StyleMilusheva, Miglena, Vera Gledacheva, Iliyana Stefanova, and Stoyanka Nikolova. 2023. "1-(2-Chlorophenyl)-6,7-dimethoxy-3-methyl-3,4-dihydroisoquinoline" Molbank 2023, no. 2: M1608. https://doi.org/10.3390/M1608
APA StyleMilusheva, M., Gledacheva, V., Stefanova, I., & Nikolova, S. (2023). 1-(2-Chlorophenyl)-6,7-dimethoxy-3-methyl-3,4-dihydroisoquinoline. Molbank, 2023(2), M1608. https://doi.org/10.3390/M1608