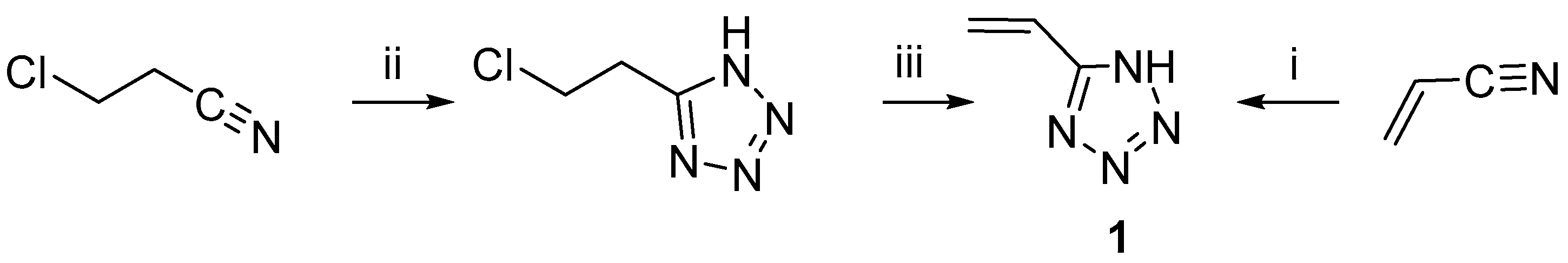
2.1. Synthesis of 5-Vinyl-1H-tetrazole
First, 5-vinyltetrazole
1 was synthesized by Arnold C. and Thatcher D. from acrylonitrile or 5-chloroethyltetrazole according to
Scheme 1 [
9].
The described methods did not spread widely in use: compound
1, which was obtained under the conditions specified by the authors, is practically impossible to purify from polymer impurities [
9]. Furthermore, aluminum oxide and hydrogen azide—byproducts in situ in the reaction mixture—are highly explosive and extremely toxic.
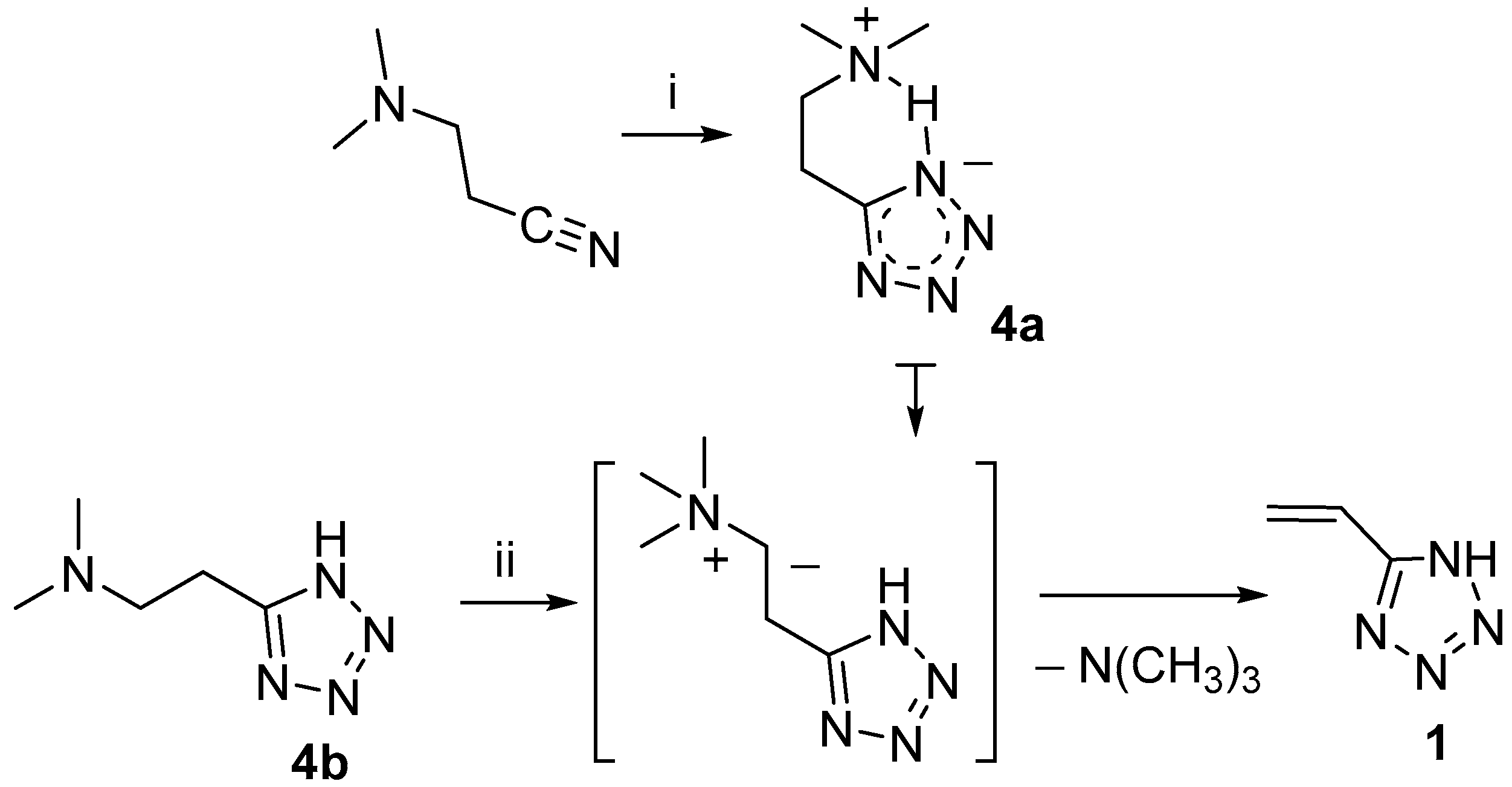
In this work, the synthesis of 5-vinyl-1
H-tetrazole
1 was based on the two-stage method, which was published earlier by our scientific group [
10]. In the first stage, as a result of 1,3-dipolar cycloaddition of dimethylammonium azide to β-dimethylaminopropionitrile in DMF, 5-(β-dimethylaminoethyl)tetrazole with a zwitter-ion structure
4a was obtained, which precipitated [
11]. In the second stage, exhaustive alkylation in H
2O of the terminal dimethylamino group of 5-(β-dimethylaminoethyl)tetrazole in normal form
4b was carried out selectively, without affecting the tetrazole ring [
10]. According to the mechanism established in work [
12], trimethylammonium salt undergoes the elimination of trimethylamine with regeneration of the double bond (Hofmann elimination) in position 5 of the tetrazole ring, with the formation of 5-vinyl-1
H-tetrazole (
Scheme 2) [
10]. This exact method is what we improve in the current work by introducing important minor adjustments for enhancing the quality of the product. As seen from
Scheme 2 and following the synthesis method, we included an additional purification step for 5-β-dimethylaminoethyltetrazole with activated charcoal. Additionally, in the method used in [
10], the obtained 5-vinyl-1
H-tetrazole was dissolved in chloroform at the boiling point of the solvent (61.2 °C) before crystallization, which often leads to the spontaneous polymerization of the product and increases its impurity. According to these circumstances, we dissolved 5-vinyl-1
H-tetrazole
1 at a temperature not exceeding 50 °C before crystallization from chloroform.
2.3. X-ray Diffraction Analysis and Quantum Chemical Calculations
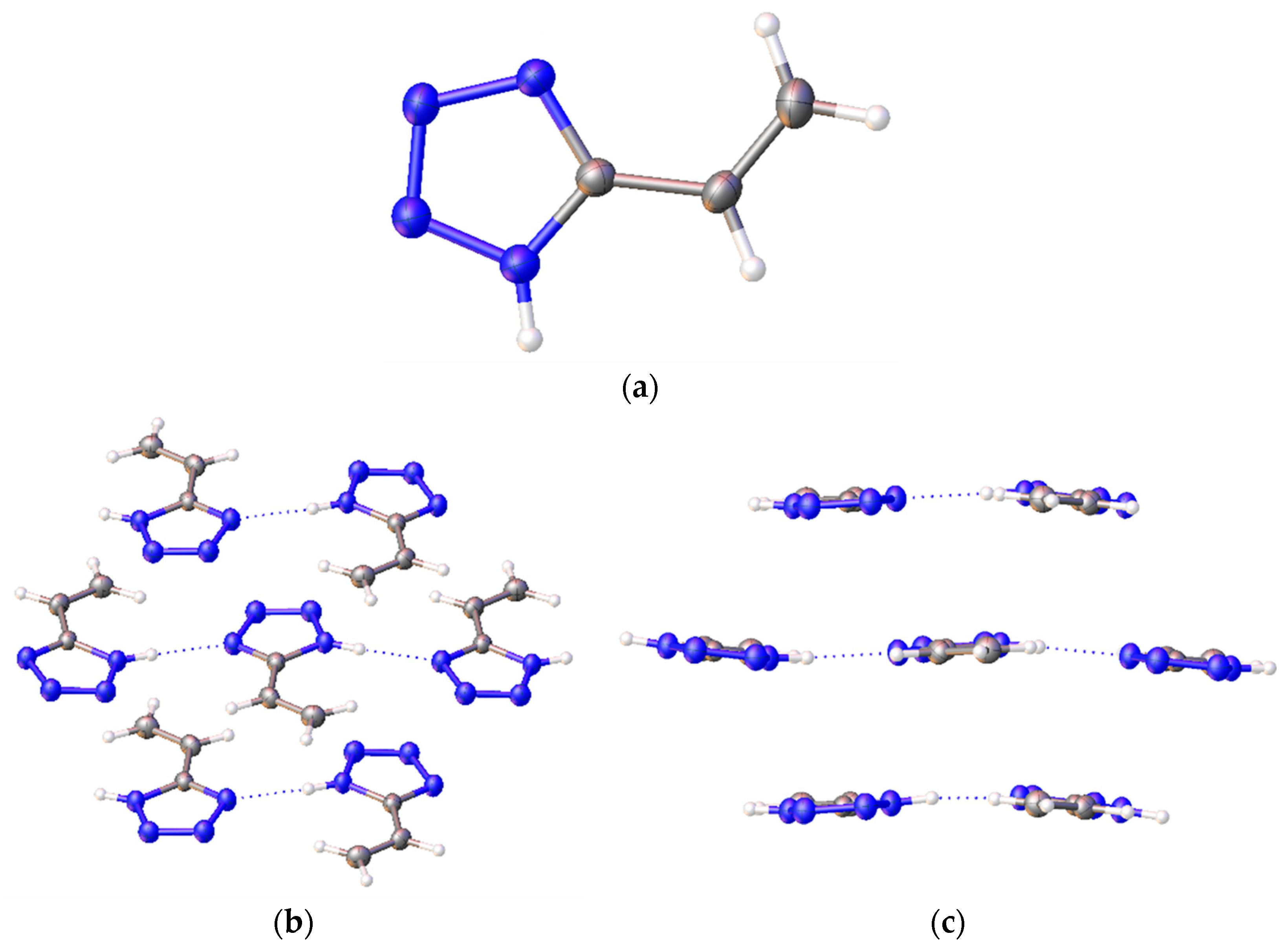
In the crystal (
Figure 3), the molecules of 5-vinyl-1
H-tetrazole
1 are held by a pair of NH···N hydrogen bonds, thus forming an infinite chain. In the lateral projection, these chains line up in almost flat plates. The distance between these plates varies and is about 3.2 Å.
The results of the X-ray diffraction of compound
1 are shown in
Table 1. The crystals are monoclinic with dimensions of 0.1 × 0.06 × 0.03 mm
3.
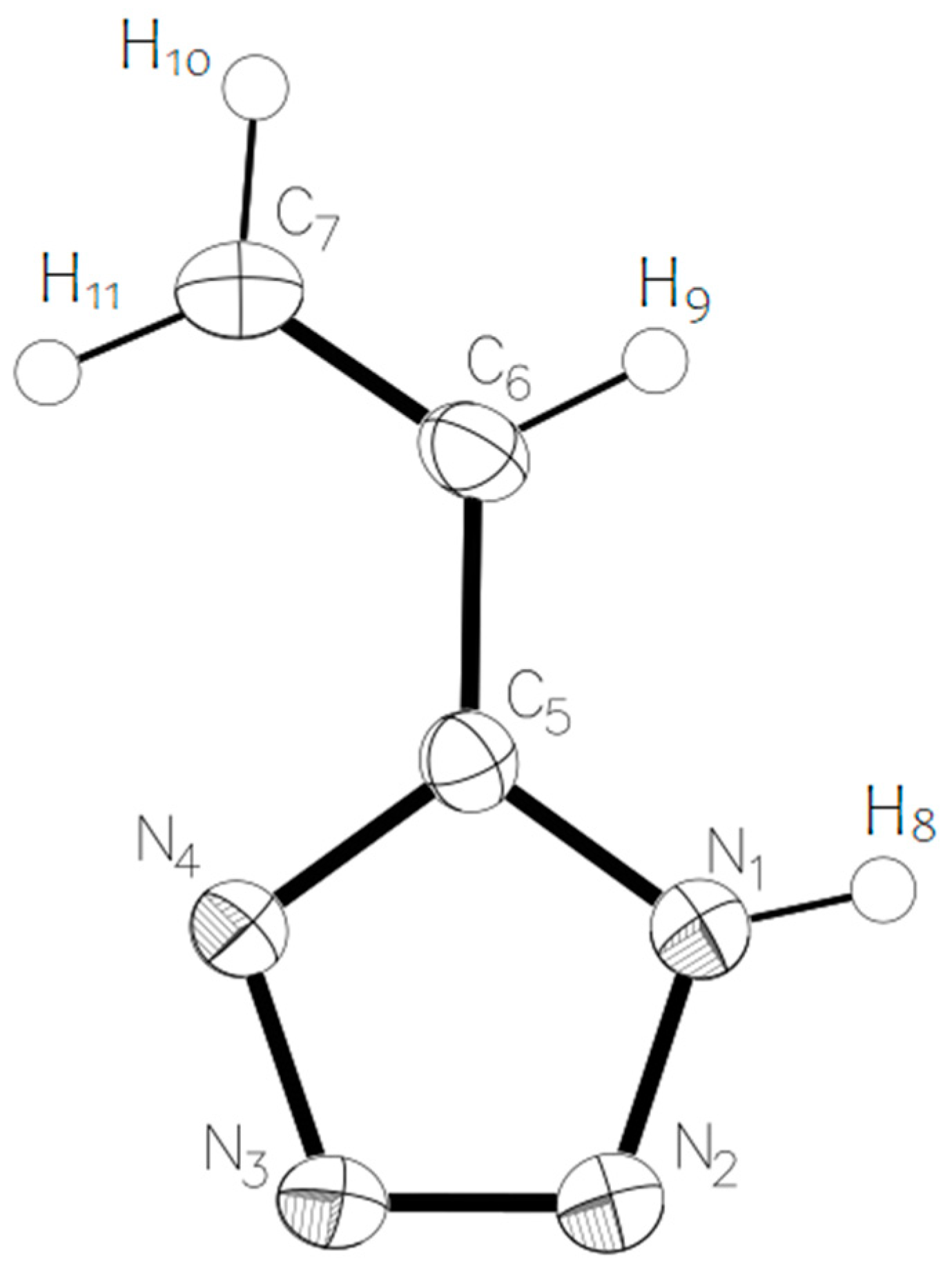
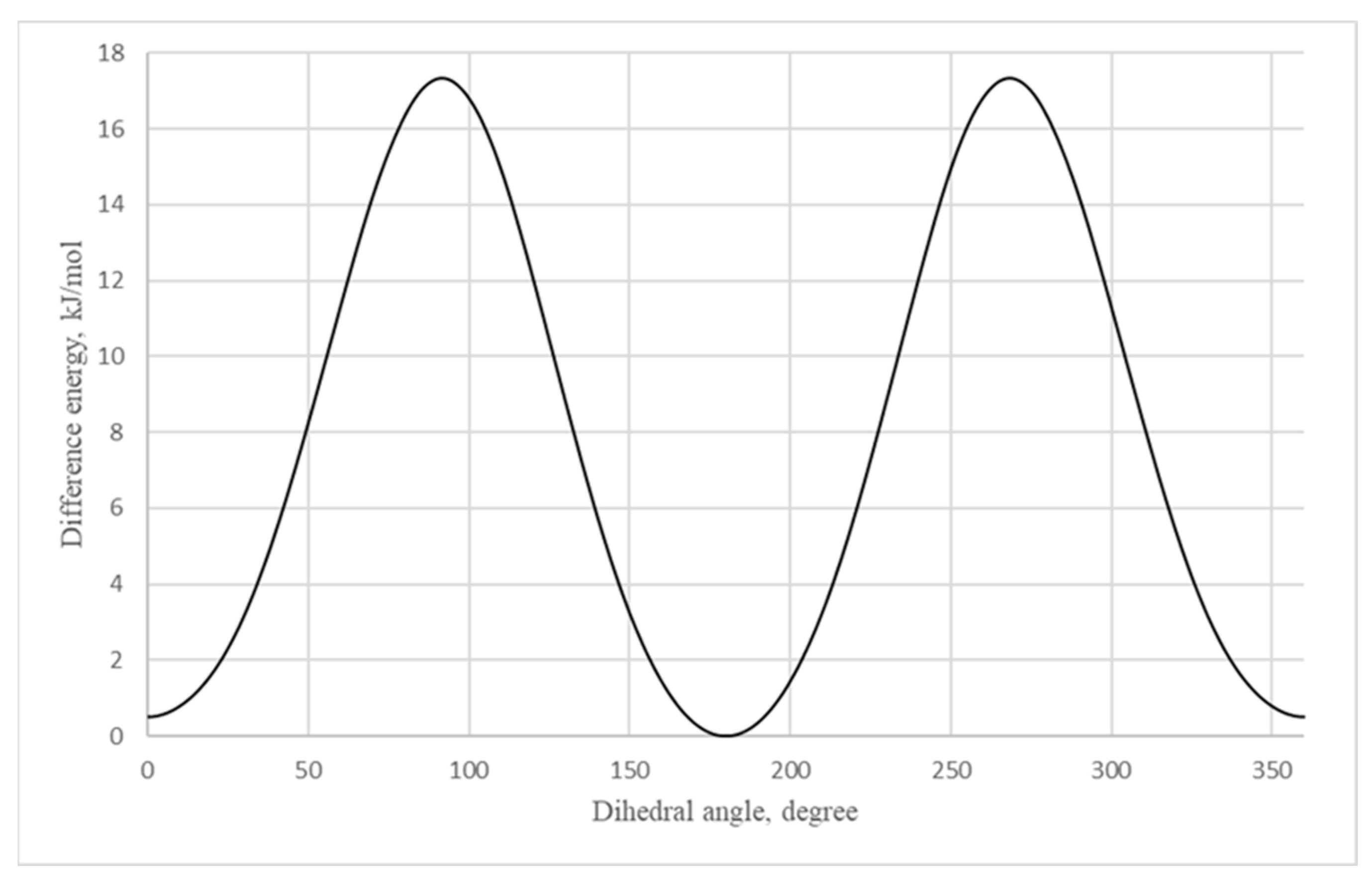
For theoretical calculations, the potential energy surface near the dihedral angle N4-C5-C6-C7 was scanned (the molecular model of vinyltetrazole with atomic numbering is shown in
Figure 4). On a graph that describes the dependence of the potential energy of vinyltetrazole on the value of the dihedral angle N4-C5-C6-C7 (
Figure 5), the energy of the most stable isomer was taken as zero, and the zero value of the dihedral angle corresponds to the structure of the molecule, as shown in
Figure 4.
According to the calculation, the 1H-5-vinyltetrazole form has to be more stable in the gas phase with 0.5 kJ/mol more energy. The results of the X-ray diffraction show that a 4H- form of the molecule is prevalent in the crystal. This may be due to the presence of intramolecular cooperation: the occurring molecular energy of the 4H- form is greater than that of the 1H- form. However, it should be expected that there is a dynamic equilibrium between the two forms of the molecule’s existence in a crystalline phase. The rotation barrier of the vinyl fragment is 17.3 kJ/mol.
The calculated quantum chemical and experimentally detected geometrical parameters of 5-vinyl-1
H-tetrazole
1 are shown in
Table 2 (with the numbering of atoms displayed in accordance with
Figure 4).
The deviation between the calculated and experimental parameters is up to two-hundredths of an angstrom for the bond lengths and up to five degrees for the bond angles. Such a deviation may be connected with the fact that the calculations were performed for an isolated molecular system, whose geometrical parameters are fundamentally different from the parameters of a molecule in a crystal.
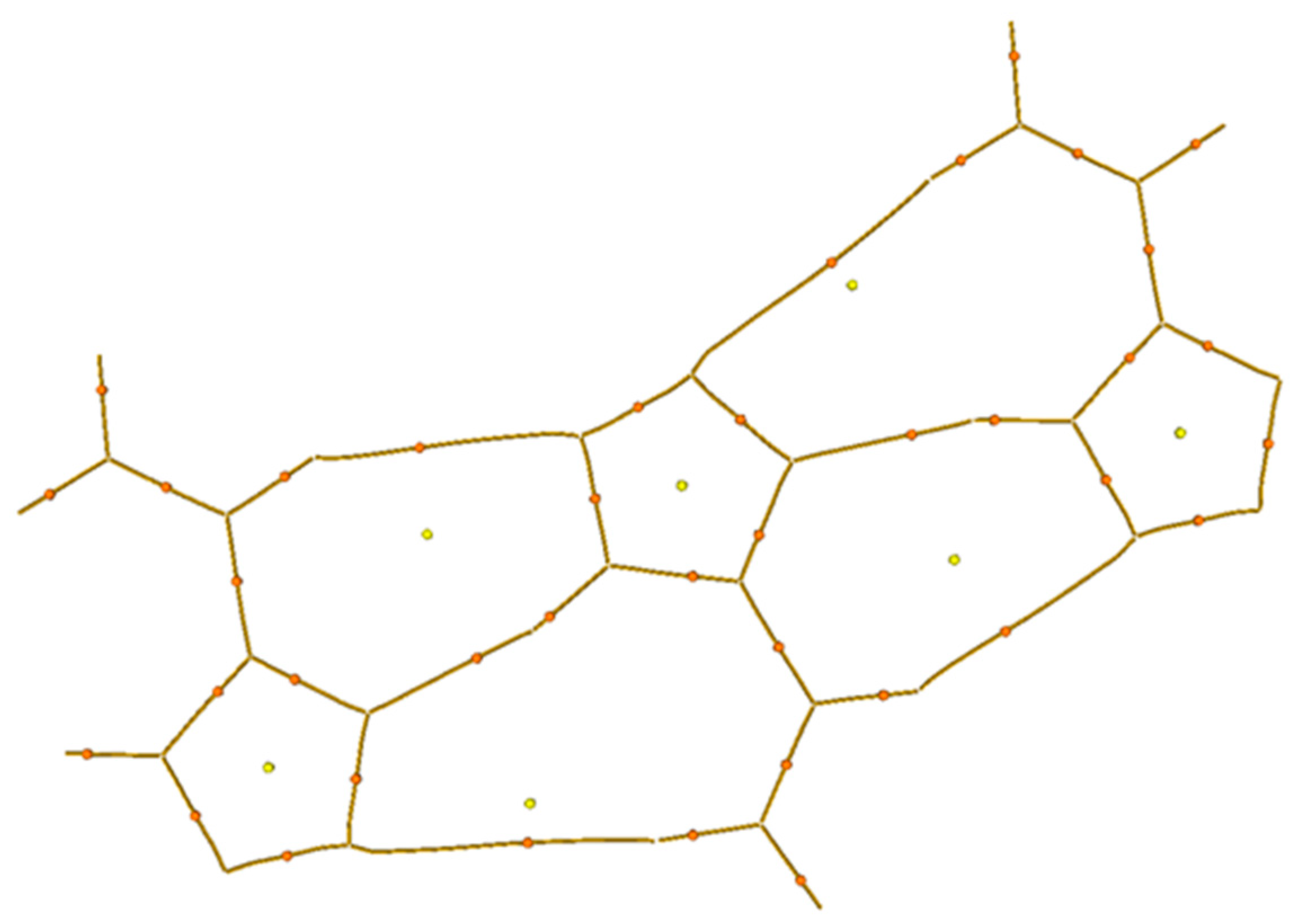
A topological analysis of the electron-density distribution in the three-molecule system was carried out (
Figure 3c, in the middle). This type of analysis facilitates the recognition of critical bond points, which are defined as extrema of electron density. Critical points were assigned as p rank, equal to the number of non-zero eigenvalues of the Hess matrix, and as q rank, equal to the algebraic sum of the signs with non-zero eigenvalues. Thus, a set of four types of critical points was obtained: points (3; −3) correspond to the nucleus position, points (3; −1) are between chemically bonded atoms, points (3; +1) are inside a flat circle and (3;+3) are inside a three-dimensional closed structure. Such an analysis, among other things, reveals the presence of hydrogen bonds in organic compounds [
13].
On the basis of the analysis results, a molecular graph using lines that connect critical points (3; −3) and (3; −1) was constructed (
Figure 6). As is seen from the graph, between the hydrogen atoms of one molecule and the nitrogen atoms of another molecule, a critical point of bond is observed. This means that a hydrogen bond exists in such a system, which corresponds with the results of the X-ray diffraction analysis. These critical point parameters and the characteristics of the nitrogen–hydrogen bond in the system with one molecule are presented in
Table 3. The Laplacian electron density values ∇²(ρ) show that the discussed intramolecular bonds have a different nature than the N-H covalent bonds with a Laplacian value of about −1.7. A similar result is shown in the value of the electron density ρ, which is greater than 0.3 for covalently bonded atoms, while for the discussed hydrogen bonds it is 0.003–0.037. The ellipticity parameter ε remains approximately the same for all the given bonds, but for the N2-(H-C) bond it sharply increases to 1.137, which is most likely because of the interaction of the nitrogen atom with the pi electrons of the carbon–carbon double bond.
In general, the results of AIM analysis indicate the existence of hydrogen bonds. However, it is noteworthy that for N-(H-C) bonds, the density of the H energy has a positive value, which means that atom repulsion takes place in such an interaction. For this reason, these interactions are not hydrogen bonds, unlike bonds of the N—(H-N) type for which both the electron density is higher and the value of the energy density at the critical point is negative.
To describe the electron-density distribution, the charges of the atoms and the indices for the one- and three-molecule systems were calculated. The results are presented in
Table 4. The calculations were made according to the theories of atoms in molecule (AIM) and natural bond orbitals (NBO). The table shows the charges of the atoms according to the definitions by Bader (AIM—charges) [
14] and by Weinhold (NBO charges) [
15]. The bond order according to the Laplacian indices for AIM analysis and Wiberg bond indices for NBO analysis were used as bond-multiplicity indices.
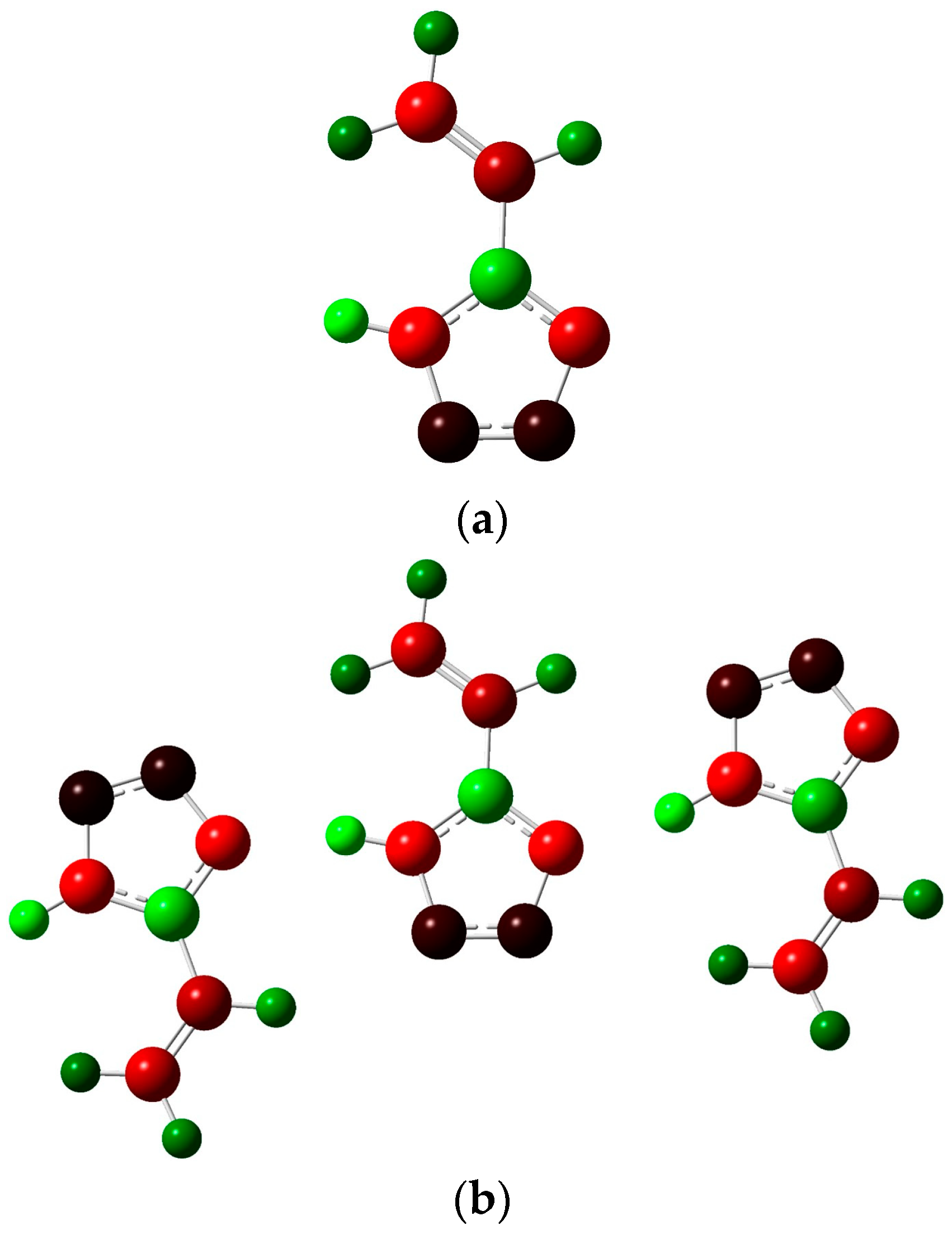
The NBO charges of the atoms are represented for one molecule (
Figure 7a) and for a system of three molecules (
Figure 7b).
On the whole, the nature of a charge distribution corresponds to general chemical concepts. It could be noticed that the N4-H8 bond in the three-molecule system weakens compared to the single molecule, which is associated with hydrogen bond formation. The Laplacian bond order for such hydrogen bonds is about 10−4, while the Wiberg bond index for them is about 0.07. This indicates the low energy of such interactions, which is consistent with the characteristics of the critical points given above.
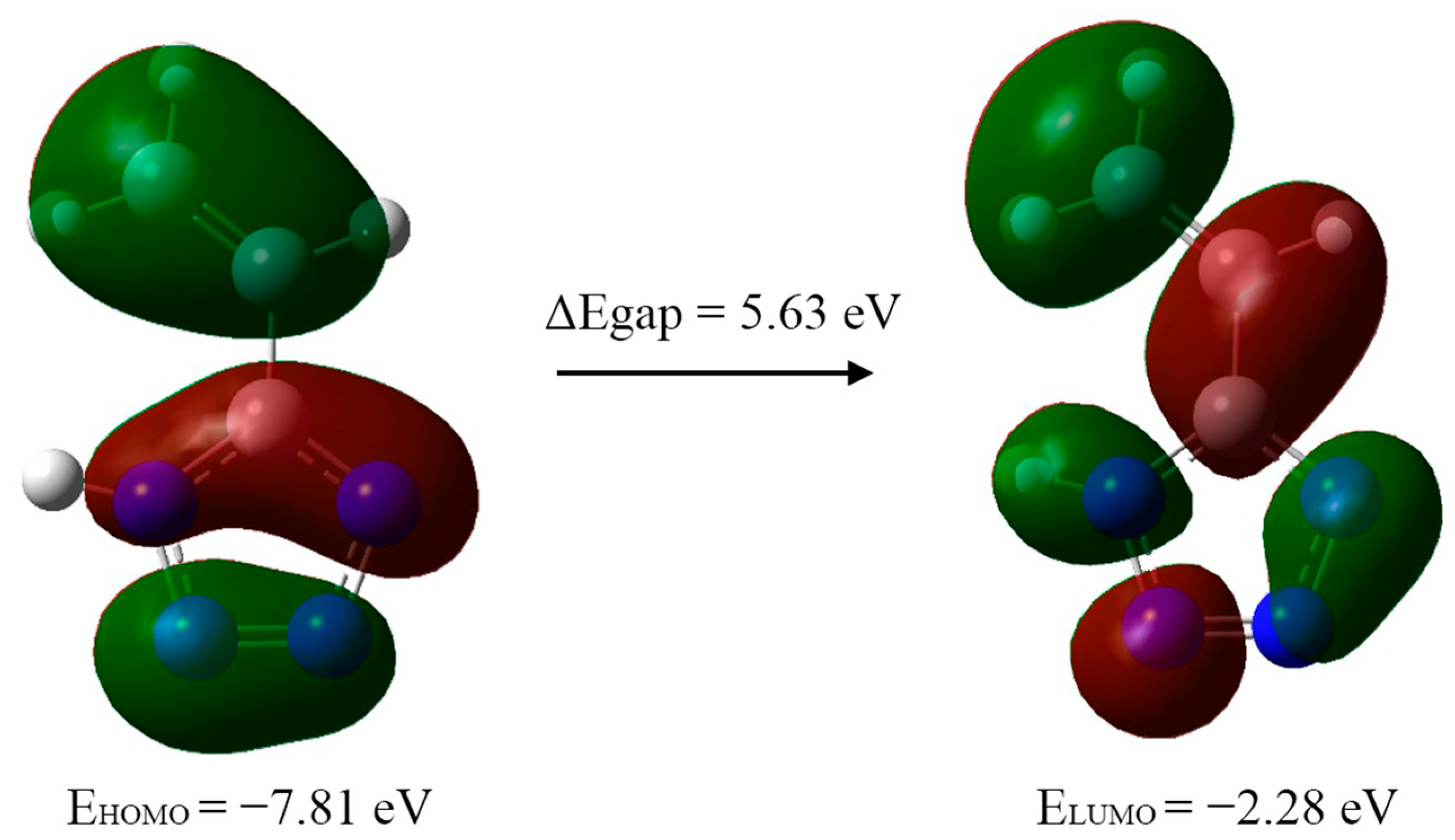
To provide a description of the electron-density distribution and chemical properties of 5-vinyl-1
H-tetrazole, the energy levels of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) were calculated (
Figure 8). The energies of these orbitals correspond with the ability to donate and accept electrons, respectively.
On the basis of the HOMO and LUMO energies, the reactivity indices were calculated: io5.nization potential (IP = −E
HOMO), electron affinity (EA = −E
LUMO), electrophilicity index (ω = μ
2/2η), chemical potential (μ = −(IP + EA)/2), electronegativity (χ = (IP + EA)/2) and hardness (η = IP − EA) (
Table 5). The calculation was performed on the basis of Koopmans’ theorem, which shows that the HOMO and LUMO energies are practically equal to the ionization potential and electron affinity, respectively. The energy difference between HOMO and LUMO, the so-called energy gap, is an indicator of the stability and reactivity of the compound [
16] and is used for the description of organic compound reactivity [
17]. In this case, its value indicates the stability of the synthesized 5-vinyl-1
H-tetrazole.
To describe the reactivity, we also calculated the values of the Fukui functions
f+ [
16], which are equal to the difference in electron density at a given point between the system with N + 1 number of electrons and the initial system with N number of electrons. A visualization of the obtained results is shown in
Figure 9. Areas with a positive Fukui function value are green-colored, and areas with a negative Fukui function value are blue-colored. Thus, in the green areas, an additional electron is predominantly distributed. A nucleophilic attack will predominantly occur in the same areas because a nucleophile is an electron-rich system.
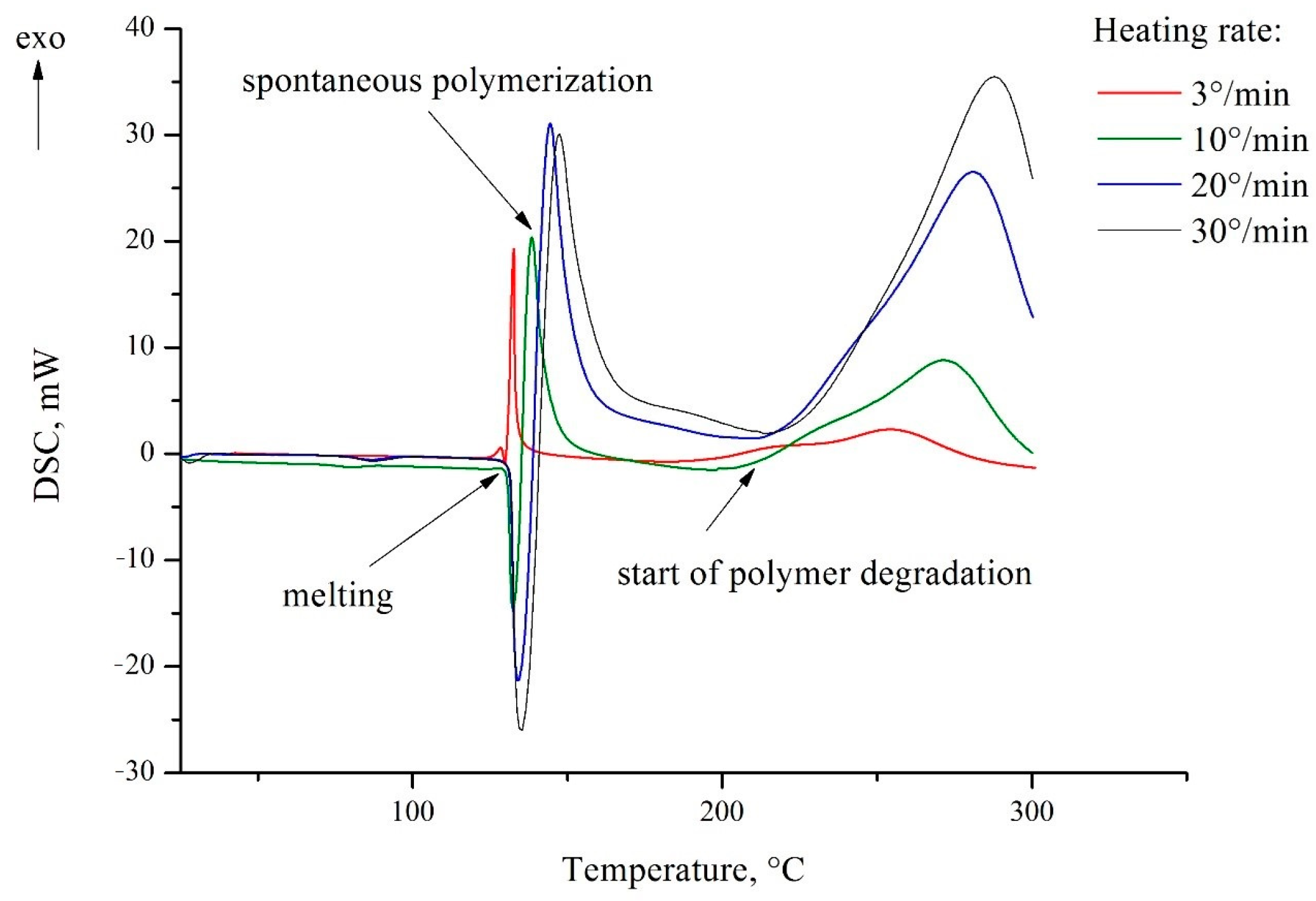
2.4. Infrared Spectroscopy
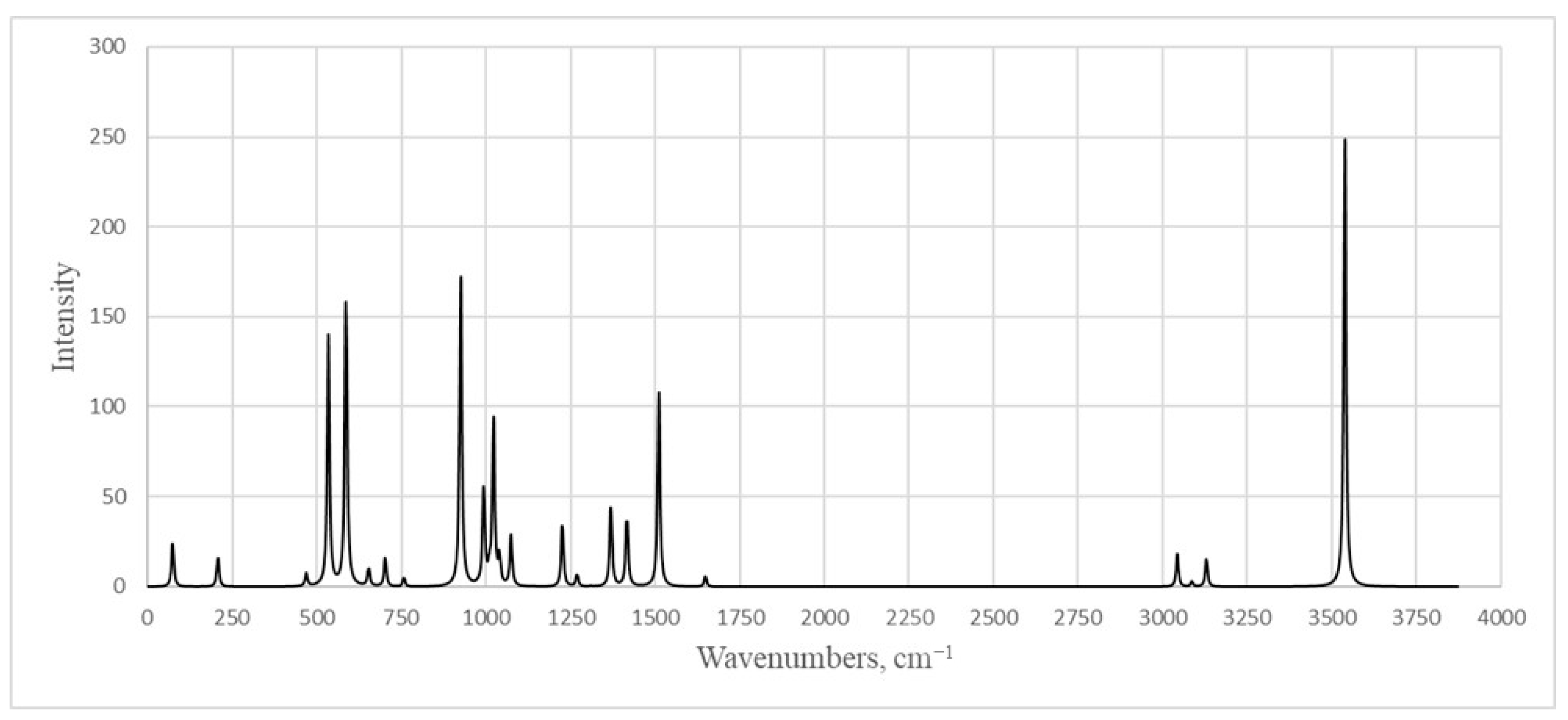
It is worth noting that the signal of an intense band corresponding to vibrations of the N–H bond at 3500 cm
−1 is absent (
Figures S2 and S3). Taking this into account, those spectra were inferred to be the crystals of 5-vinyl-1
H-tetrazole
1. Analyzing the results of the X-ray diffraction analysis and quantum chemical calculations, we can assume that the reason is the presence of the proton of the cycle in the composition of the linear associates –N-1–H···N-4–, where nitrogen atoms belong to the cycles of neighboring molecules. This fact was highlighted earlier as the characteristic feature of the crystals of all N
H-unsubstituted tetrazoles [
18]. Then, the vibration of the proton is consistent; for example, the stretching of the N-1–H bond is accompanied by the contraction of the H···N-4 bond and vice versa. This is accompanied by a shift in the resonant frequency to the lower wave number area. Interestingly, in the IR spectrum of the N-vinyl pyrrolidone and 5-vinyl-1
H-tetrazole
1 copolymer sample, which contained only 5 mol% of 5-vinyltetrazole units, a band appeared at 3500 cm-1 (
Figure S4). Because the spectrum of N-vinylpyrrolidone has no absorption in this region (
Figure S5), a band corresponds to the calculated related vibrations of the N–H bond. The difference between the polymer and monomer crystals is that the tetrazole rings were linked to the polymer chain and depended on its molecular dynamics. This makes it impossible to form associates similar to those found in the crystals of the 5-vinyl-1
H-tetrazole
1 itself.
The above reasoning is confirmed by the results of quantum chemical calculations. In the calculated IR spectrum for the three-molecule system, there are 2 peaks: 3531 cm
−1 corresponds to stretching vibrations of the N-H side molecule, and 3075 cm
−1 (the intense band) corresponds to stretching vibrations of H atoms bound by hydrogen bonds. Apparently, a corresponding vibration occurs in the experimental spectrum at the peaks of 3116 cm
−1 (
Figure S2) and 3114 cm
−1 (
Figure S3). A large difference between the experimental and calculated intensity of the discussed peaks is explained by the fact that for the vibrations that appear in the IR spectrum, the intensity is proportional to the change in the dipole moment for a given vibration. At the same time, with the scaling of a system’s size (in this case, an increase in the number of links in the chain), the dipole moment stabilizes and depends less and less on the vibration of the atom, which leads to a decrease in the vibrations’ intensity. Vibrations were observed in both the experimental and theoretical spectra (theoretical value is given in parentheses): 674.1 (653.3) cm
−1 corresponds to deformation vibrations of the vinyl fragment; 781.2 (757.5) cm
−1 corresponds to torsional vibrations of both the vinyl fragment and the ring; 935.5 (925.4) cm
−1 corresponds to torsional vibrations of CH
2; 962.5 (975.4) cm
−1 corresponds to bending vibrations of the ring; 1050.3-1248.0 cm
−1 peaks refer to stretching vibrations of the tetrazole ring; the 1371.45 (1369.2) and 1559.51 (1510.5) cm
−1 peaks correspond to deformation symmetric CH-CH
2 vibrations (in this case, the second peak corresponds to a vibration with a large amplitude of the endocyclic carbon atom); the 1645.3 (1648.3) cm
−1 peak corresponds to stretching vibrations of the vinyl fragment; and the 2872.13 (3042.3), 2924.2 (3085.3) and 2998.5 (3128.4) cm
−1 peaks refer to stretching vibrations of hydrogen atoms in the vinyl fragment. It should be noted that we used a scaling multiplier of 0.9688 for the entire range of wave numbers, and in the region of the small wave numbers, the theoretical values are somewhat underestimated compared to the experimental ones. Peaks at about 1000–1300 cm
−1 converge well, and peaks in the region above 2000 cm
−1 are strongly overestimated. Peaks that are not indicated above and that are absent from both the calculated spectra for the system of one molecule and for the system of three molecules (in the region of 2200–2850 cm
−1) were most likely interplanar vibrations in the vinyltetrazole crystal.
2.5. Mass Spectrometry
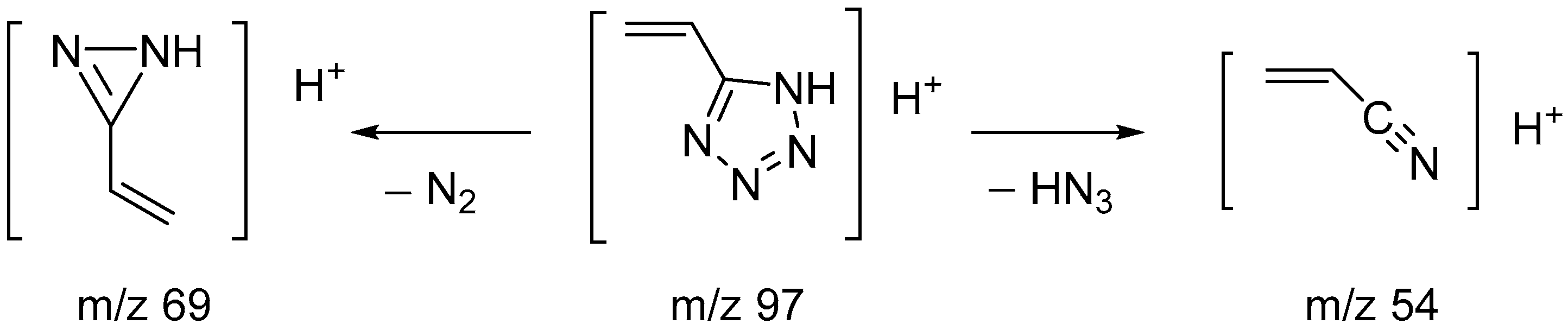
In order to confirm the structure of the synthesized compound and study the pathways of mass spectrometric fragmentation, high-resolution mass spectra in full-scan mode and tandem spectra for the protonated molecular ion were obtained. In the spectra recorded using electrospray mass spectroscopy (ESI-MS) in positive mode, a molecular ion with m/z 97.05087 was detected. Its mass differed from the theoretical (calculated) mass of 5-vinyl-tetrazole by 0.23 ppm, which fit into the mass measurement accuracy criterion (5 ppm) when identifying compounds using an LTQ Orbitrap high-resolution mass spectrometer.
The fragmentation of the molecular ions of the studied substance was strongly influenced by the collision energy in the fragmentation cell. Thus, with an increase in the fragmentation energy, the intensity of the molecular ion signal noticeably decreased [M+H]+ (
m/
z = 97), and the intensity of the fragment ions increased (
m/
z = 54) (
Table 6).
The mass spectral fragmentation of 5-vinyl-1
H-tetrazole mainly proceeded with the elimination of HN
3 (product ion with
m/
z 54), which was also noted by Forkey and Carpenter when studying the fragmentation of tetrazole, 5-methyltetrazole and 1,5-dimethyltetrazole [
19]. At high ionization energies, the nitrogen molecule (product ion with
m/
z 69) was eliminated, which is characteristic of all tetrazole derivatives [
19] (
Scheme 3).
2.6. NMR Spectroscopy
A group of signals of protons in the vinyl group, a proton cycle in a very weak field and three signals of carbon atoms are expected to be observed in the NMR 1H and 13C spectra. The endocyclic carbon atom is visible in the weakest field (
Figures S6 and S7).
Tetrazole has only two existing tautomeric forms from the four possible tautomers: 1
H-tetrazole and 2
H-tetrazole (
Figure 11). The 1
H-tautomer predominates in the condensed phase and polar solvents, whereas 2
H-tetrazole is more stable in the gas phase [
20].
It should be noted that virtually all N
H-unsubstituted 5-substituted tetrazoles, regardless of the substituent, exist in the crystalline state as individual 1
H-tautomers [
1,
18].
According to the data of the 2D correlation spectrum
1H-
15N HMBC, the proton of the CH group, which is in the alpha position to the tetrazole ring, has two significant cross-peaks with two types of nitrogen atoms at 177.7 and 345.7 ppm, while the protons of the CH
2 group have cross-peaks with only one nitrogen atom at 177.4 ppm (
Table 7,
Figure S8). Obviously, such a picture can be observed because of the fast intermolecular exchange of protons between the nitrogen atoms in positions 1 and 4 of the heteroring, which leads to an averaging of the characteristics of the N
1 and N
4 atoms as well as N
2 and N
3. Similar effects were noted previously [
20]. The probability of tautomeric transformations of 1H/2H-tetrazole seems unlikely under these conditions [
18].



 ,
, 















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
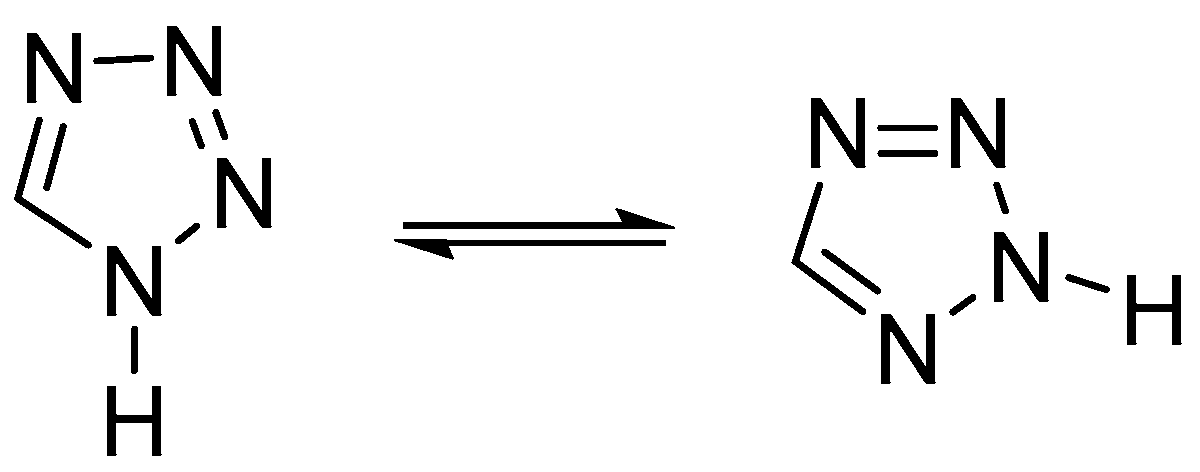{kind=link}
{kind=link}
{kind=link}
{kind=link}