
1,3,3,3′,3′-Pentaisopropyl-1,3,3′,6,6′,7,7′-heptahydro-1λ5-1,1′-spirobi[acenaphtho 5,6-cd][1,2,6]oxadiphosphinine]-3,3′-diium Triiodide


Abstract
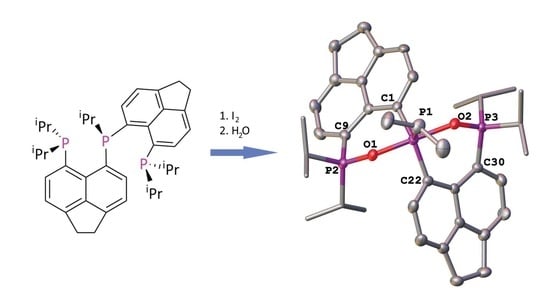
{kind=link}
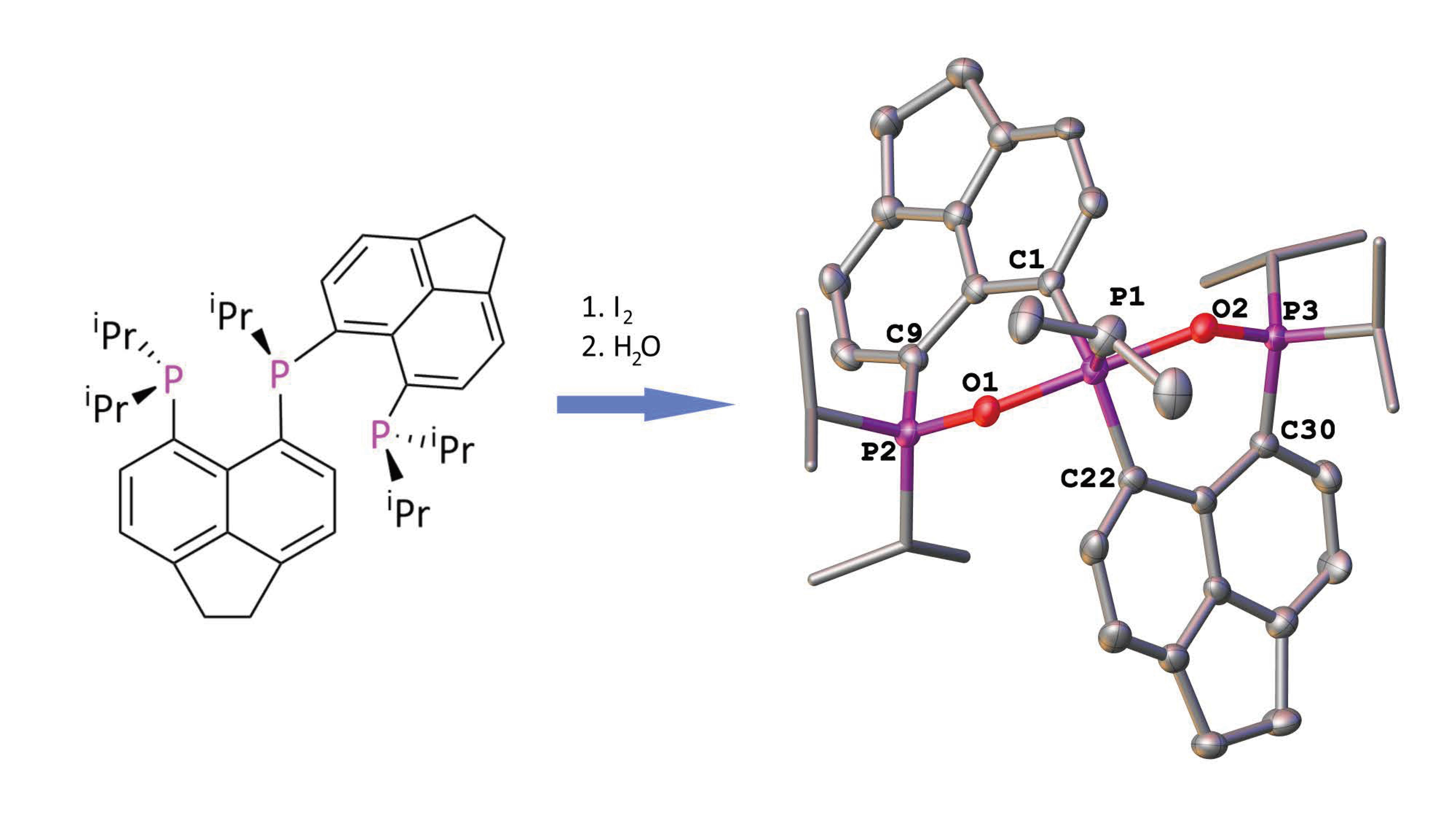
{kind=link}
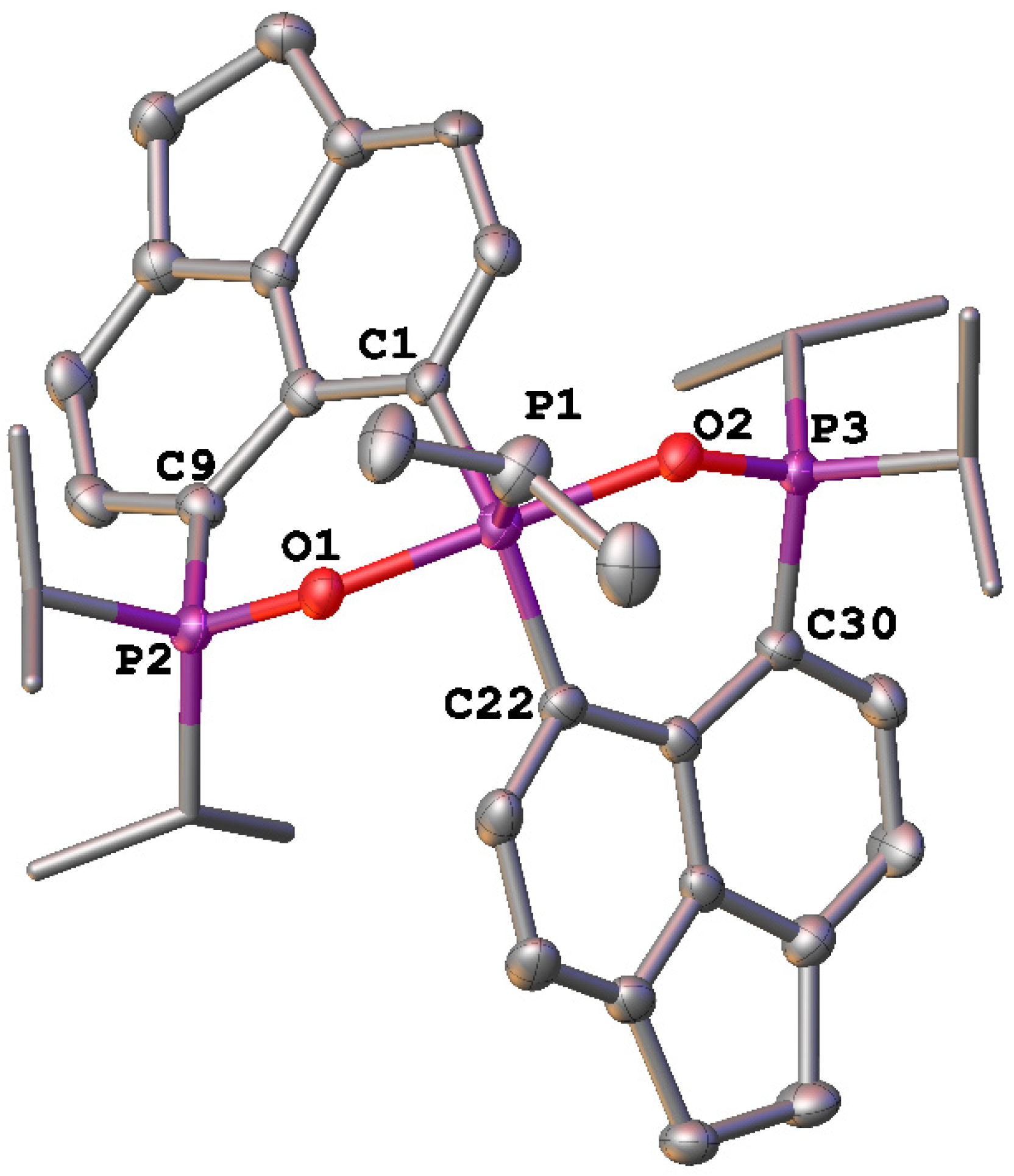
{kind=link}
{kind=link}
1. Introduction

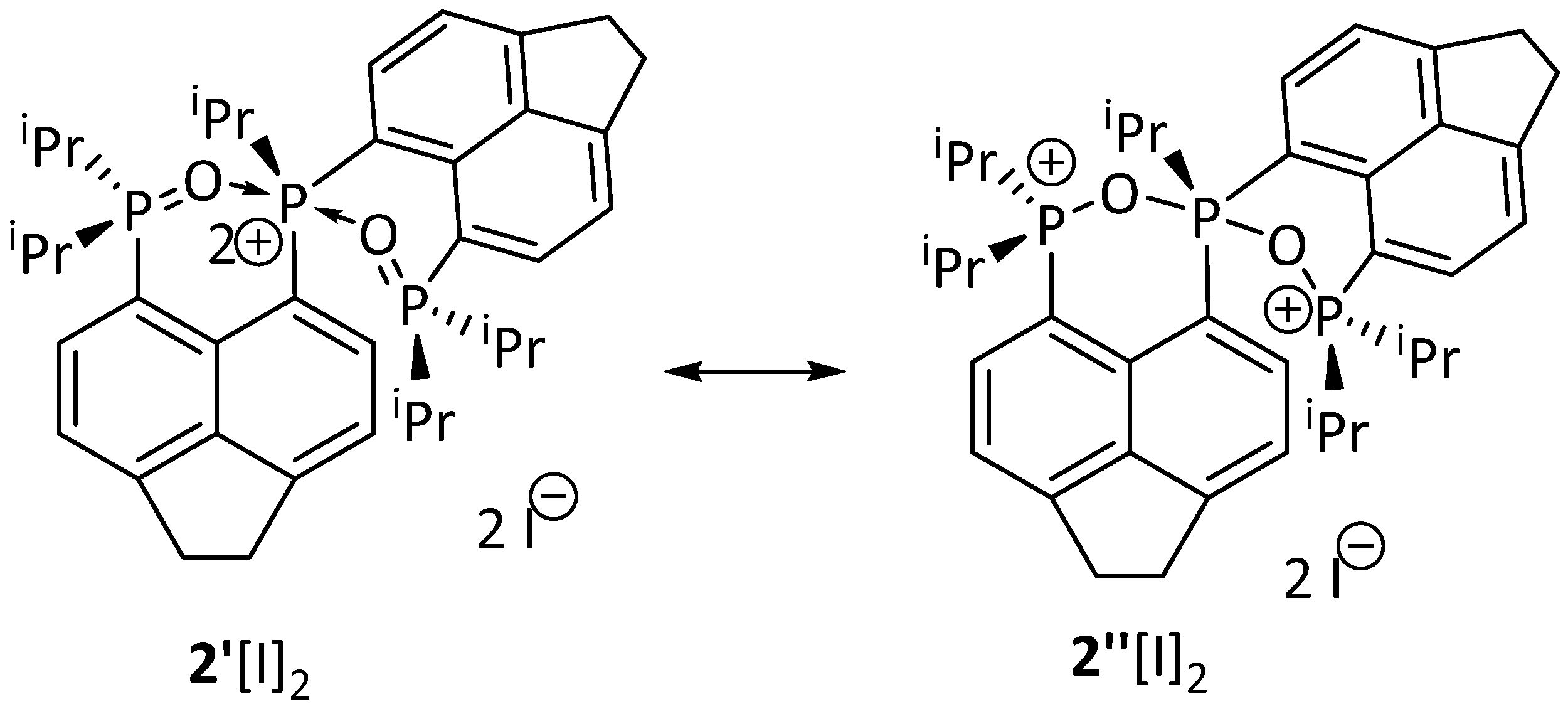
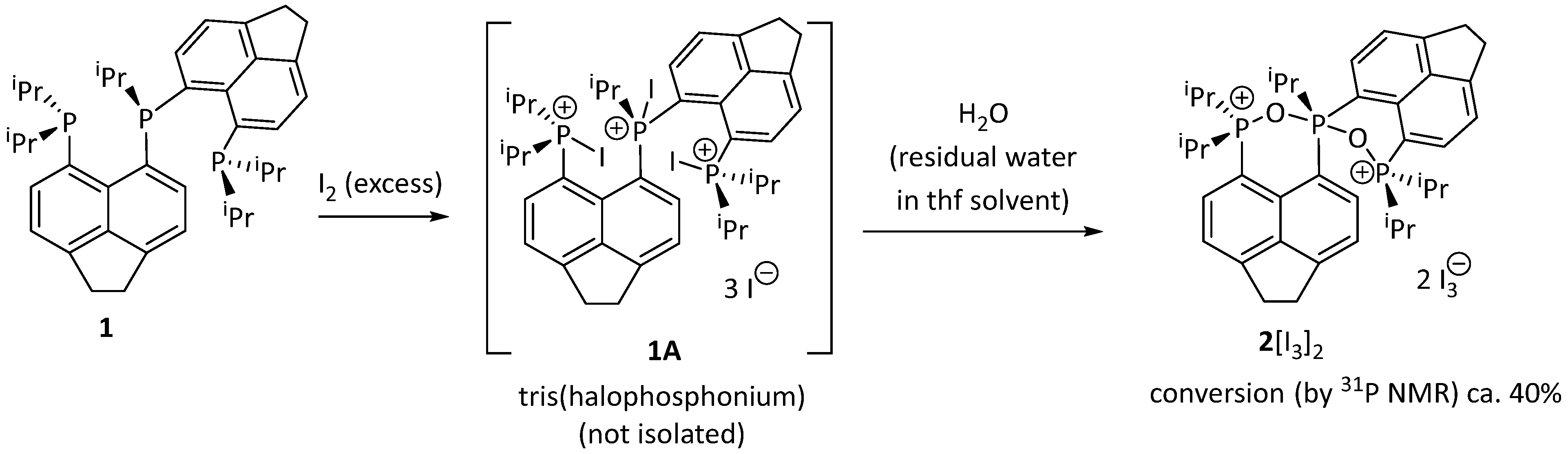
2. Results and Discussion
3. Conclusions
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Alhanash, F.B.; Barnes, N.A.; Godfrey, S.M.; Hurst, P.A.; Hutchinson, A.; Khan, R.Z.; Pritchard, R.G. Structural isomerism in tris-tolyl halo-phosphonium and halo-arsonium tri-halides, [(CH3C6H4)3EX][X3], (E = P, As; X = Br, I). Dalton Trans. 2012, 41, 7708–7728. [Google Scholar] [CrossRef] [PubMed]
- Appel, R. Tertiary Phosphane/Tetrachloromethane, a Versatile Reagent for Chlorination, Dehydration, and P—N Linkage. Angew. Chem. Int. Ed. Engl. 1975, 14, 801–811. [Google Scholar] [CrossRef]
- Quin, L.D. A Guide to Organophosphorus Chemistry; Wiley Interscience: New York, NY, USA, 2000. [Google Scholar]
- Denton, R.M.; An, J.; Adeniran, B.; Blake, A.J.; Lewis, W.; Poulton, A.M. Catalytic Phosphorus(V)-Mediated Nucleophilic Substitution Reactions: Development of a Catalytic Appel Reaction. J. Org. Chem. 2011, 76, 6749–6767. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; An, J.; Denton, R.M. A procedure for Appel halogenations and dehydrations using a polystyrene supported phosphine oxide. Tetrahedron Lett. 2014, 55, 799–802. [Google Scholar] [CrossRef]
- Xia, X.; Toy, P.H. Rasta resin–triphenylphosphine oxides and their use as recyclable heterogeneous reagent precursors in halogenation reactions. Beilstein J. Org. Chem. 2014, 10, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, K.V.; Gilheany, D.G. Identification of a key intermediate in the asymmetric Appel process: One pot stereoselective synthesis of P-stereogenic phosphines and phosphine boranes from racemic phosphine oxides. Chem. Commun. 2012, 48, 10040–10042. [Google Scholar] [CrossRef] [PubMed]
- Vetter, A.C.; Nikitin, K.; Gilheany, D.G. Long sought synthesis of quaternary phosphonium salts from phosphine oxides: Inverse reactivity approach. Chem. Commun. 2018, 54, 5843–5846. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, K.; Jennings, E.V.; Al Sulaimi, S.; Ortin, Y.; Gilheany, D.G. Dynamic Cross-Exchange in Halophosphonium Species: Direct Observation of Stereochemical Inversion in the Course of an SN2 Process. Angew. Chem. Int. Ed. 2018, 57, 1480–1484. [Google Scholar] [CrossRef] [PubMed]
- Jennings, E.V.; Nikitin, K.; Ortin, Y.; Gilheany, D.G. Degenerate Nucleophilic Substitution in Phosphonium Salts. J. Am. Chem. Soc. 2014, 136, 16217–16226. [Google Scholar] [CrossRef] [PubMed]
- Al-Farhan, K.A. Crystal structure of triphenylphosphine oxide. J. Crystallogr. Spectrosc. Res. 1992, 22, 687–689. [Google Scholar] [CrossRef]
- Chandrasekaran, A.; Day, R.O.; Holmes, R.R. Isomeric Intraconversion among Penta- and Hexacoordinate Cyclic Oxyphosphoranes via Oxygen Atom Coordination 1,2. J. Am. Chem. Soc. 1997, 119, 11434–11441. [Google Scholar] [CrossRef]
- Jiang, X.-D.; Matsukawa, S.; Yamamichi, H.; Kakuda, K.-i.; Kojima, S.; Yamamoto, Y. Stereomutation and Experimental Determination of the Relative Stability of Diastereomeric O-Equatorial Anti-Apicophilic Spirophosphoranes. Eur. J. Org. Chem. 2008, 2008, 1392–1405. [Google Scholar] [CrossRef]
- Chandrasekaran, A.; Day, R.O.; Holmes, R.R. Coordination of Carbonyl and Carboxyl Oxygen Atoms with Phosphorus in the Presence of Hydrogen Bonding. P−O Donor Action. Inorg. Chem. 2001, 40, 6229–6238. [Google Scholar] [CrossRef]
- Ray, M.J.; Randall, R.A.M.; Arachchige, K.S.A.; Slawin, A.M.Z.; Buehl, M.; Lebl, T.; Kilian, P. Synthetic, Structural, NMR, and Computational Study of a Geminally Bis(peri-substituted) Tridentate Phosphine and Its Chalcogenides and Transition-Metal Complexes. Inorg. Chem. 2013, 52, 4346–4359. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Picthall, D.; Ray, M.J.; Slawin, A.M.Z.; Kilian, P. 1,3,3,3′,3′-Pentaisopropyl-1,3,3′,6,6′,7,7′-heptahydro-1λ5-1,1′-spirobi[acenaphtho 5,6-cd][1,2,6]oxadiphosphinine]-3,3′-diium Triiodide. Molbank 2022, 2022, M1523. https://doi.org/10.3390/M1523
Picthall D, Ray MJ, Slawin AMZ, Kilian P. 1,3,3,3′,3′-Pentaisopropyl-1,3,3′,6,6′,7,7′-heptahydro-1λ5-1,1′-spirobi[acenaphtho 5,6-cd][1,2,6]oxadiphosphinine]-3,3′-diium Triiodide. Molbank. 2022; 2022(4):M1523. https://doi.org/10.3390/M1523
Chicago/Turabian StylePicthall, Daniel, Matthew James Ray, Alexandra Martha Zoya Slawin, and Petr Kilian. 2022. "1,3,3,3′,3′-Pentaisopropyl-1,3,3′,6,6′,7,7′-heptahydro-1λ5-1,1′-spirobi[acenaphtho 5,6-cd][1,2,6]oxadiphosphinine]-3,3′-diium Triiodide" Molbank 2022, no. 4: M1523. https://doi.org/10.3390/M1523
APA StylePicthall, D., Ray, M. J., Slawin, A. M. Z., & Kilian, P. (2022). 1,3,3,3′,3′-Pentaisopropyl-1,3,3′,6,6′,7,7′-heptahydro-1λ5-1,1′-spirobi[acenaphtho 5,6-cd][1,2,6]oxadiphosphinine]-3,3′-diium Triiodide. Molbank, 2022(4), M1523. https://doi.org/10.3390/M1523