
Activation–Deactivation of Inter-Peptide Bond in Fluoro-N-(2-hydroxy-5-methyl phenyl)benzamide Isomers, Induced by the Position of the Halogen Atom in the Benzene Ring

 , , ,
, , ,  and
and 
Abstract
:
1. Introduction
2. Results
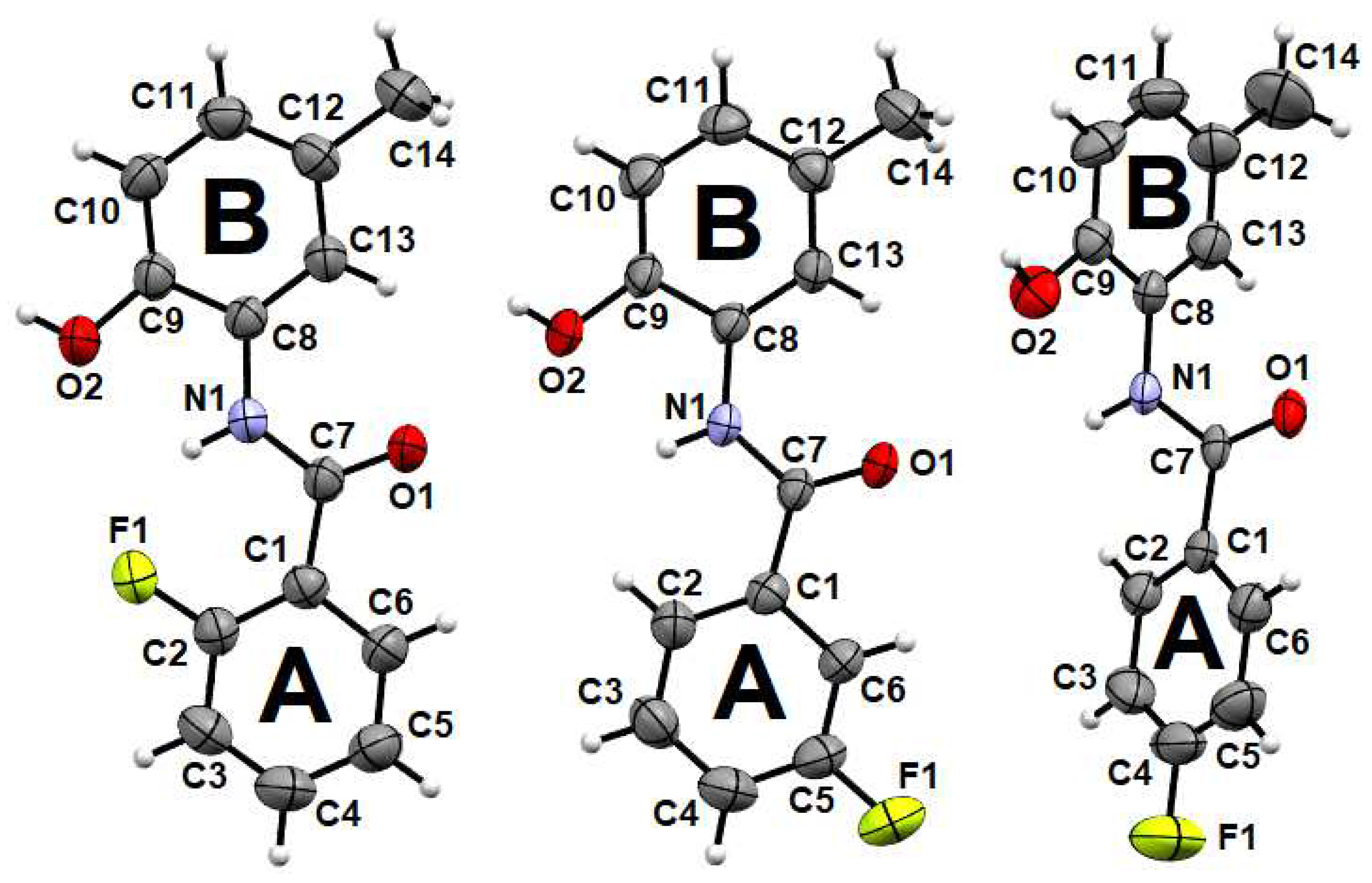
2.1. Structural Analysis
Crystal Data
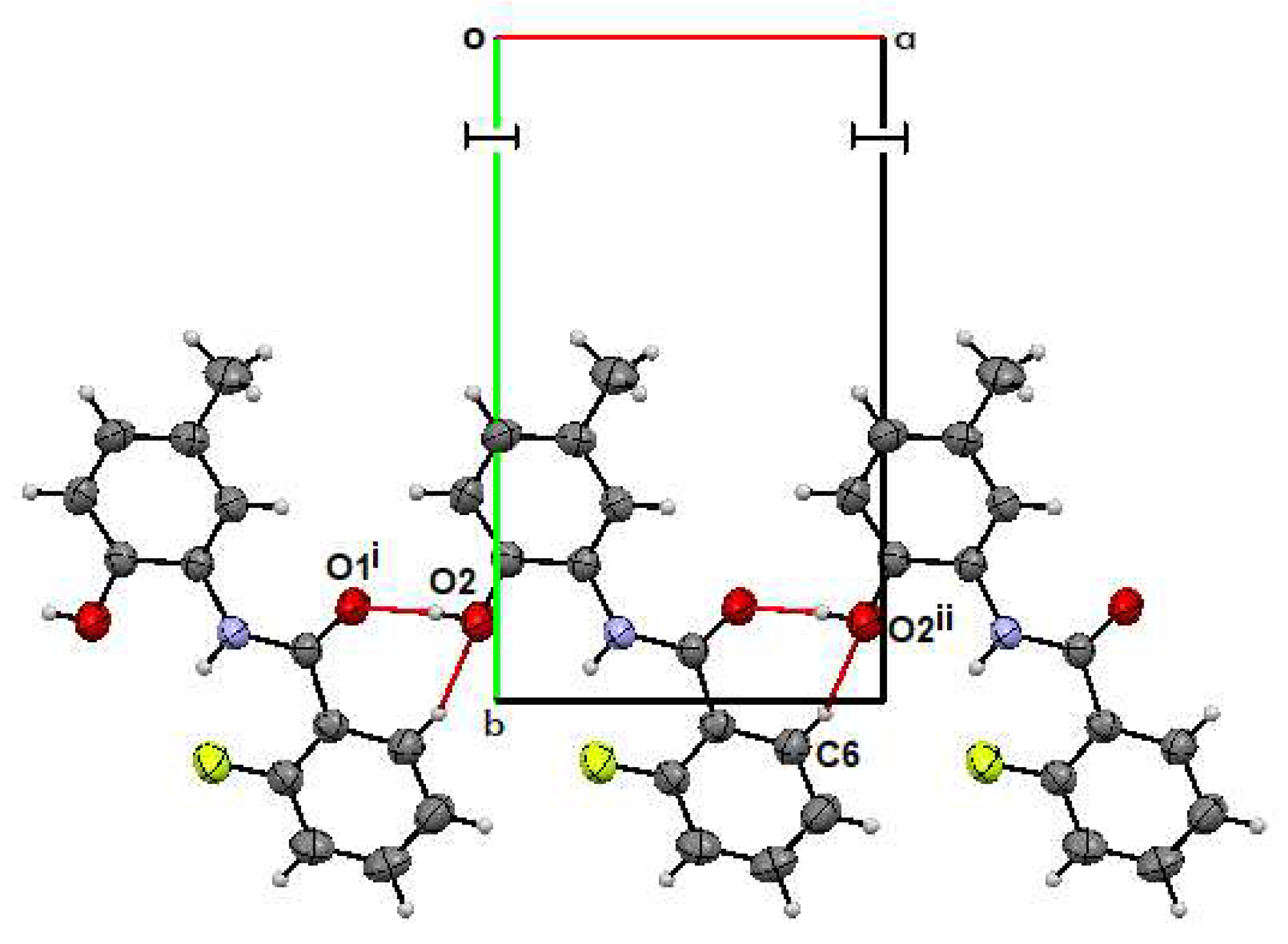
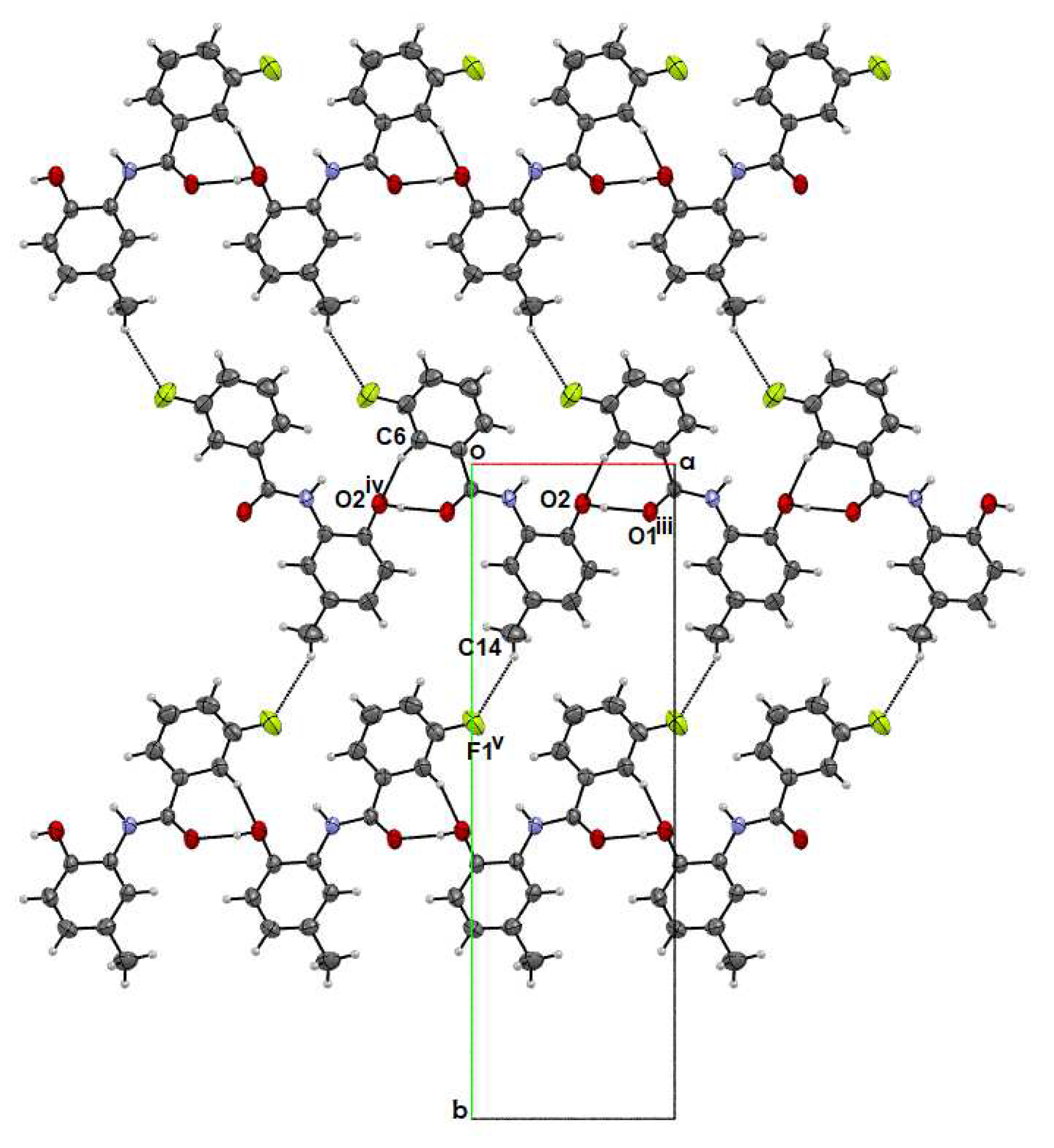
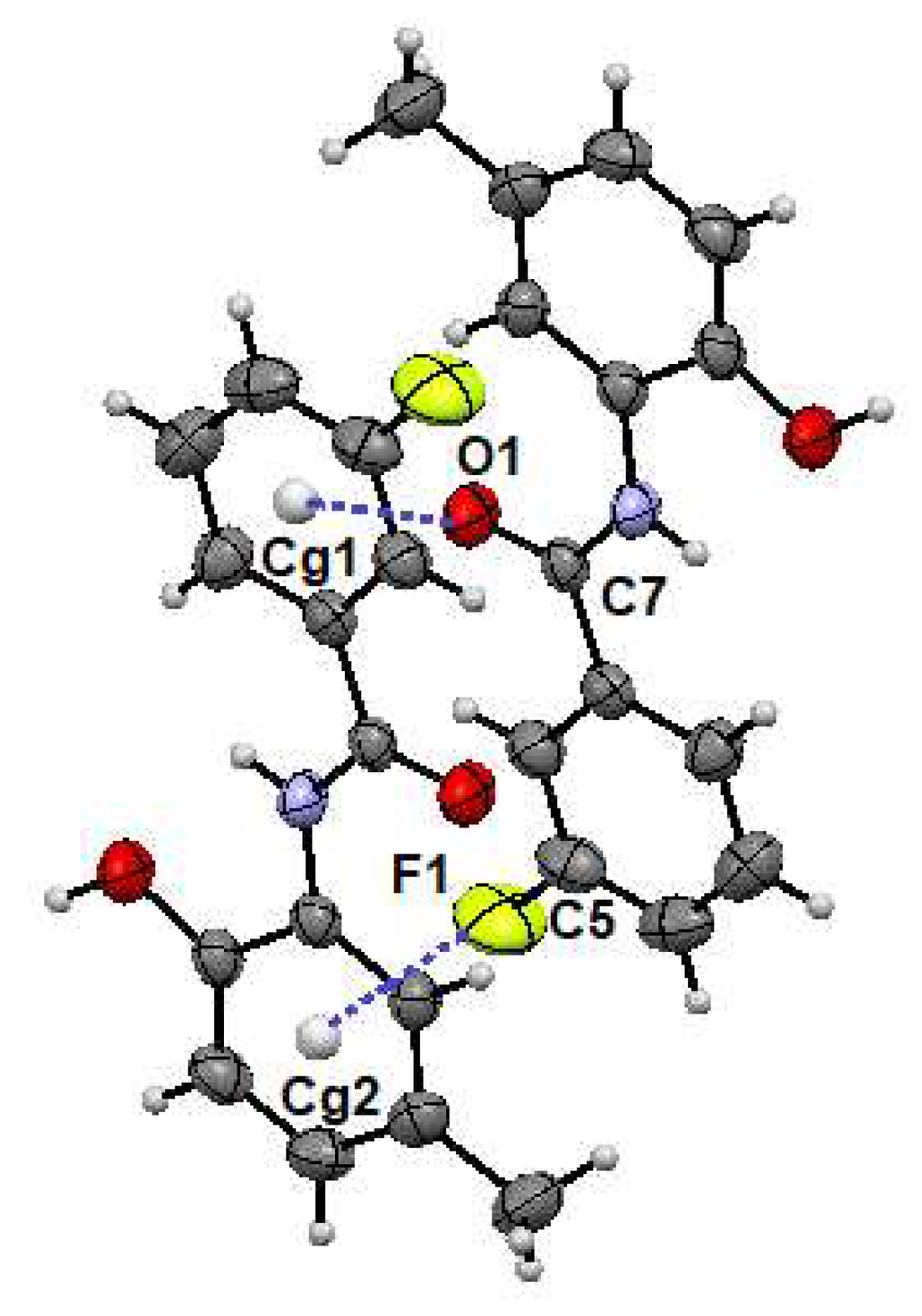
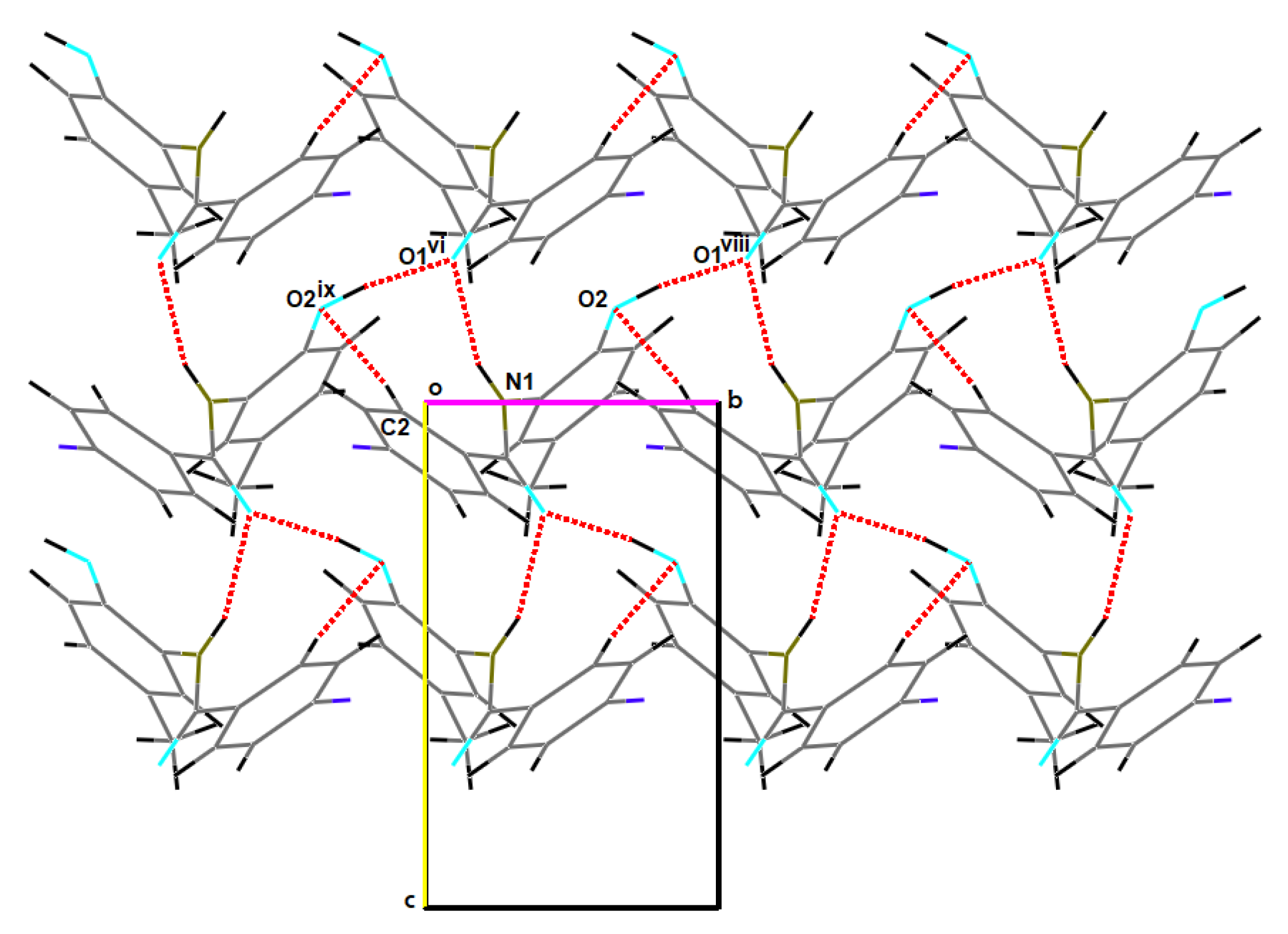
2.2. Supramolecular Features
2.2.1. o-FPhB
2.2.2. m-FPhB
2.2.3. p-FPhB
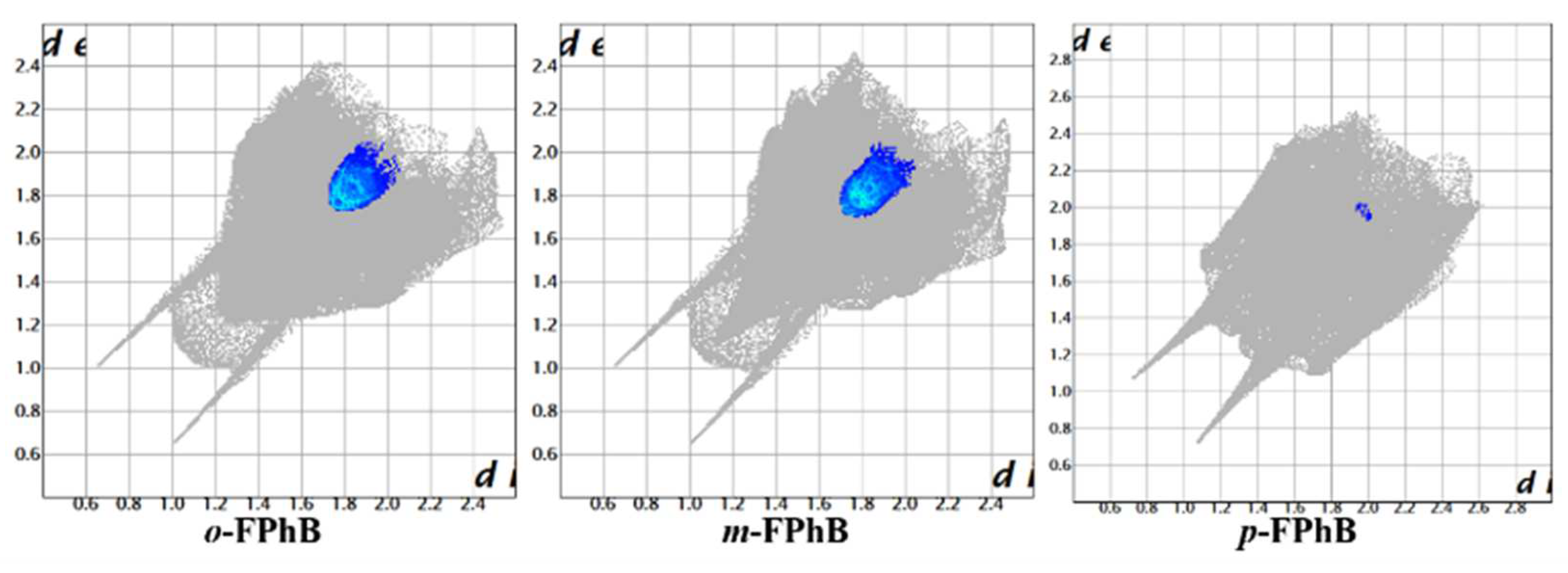
2.3. Analysis of the Hirshfeld Surfaces
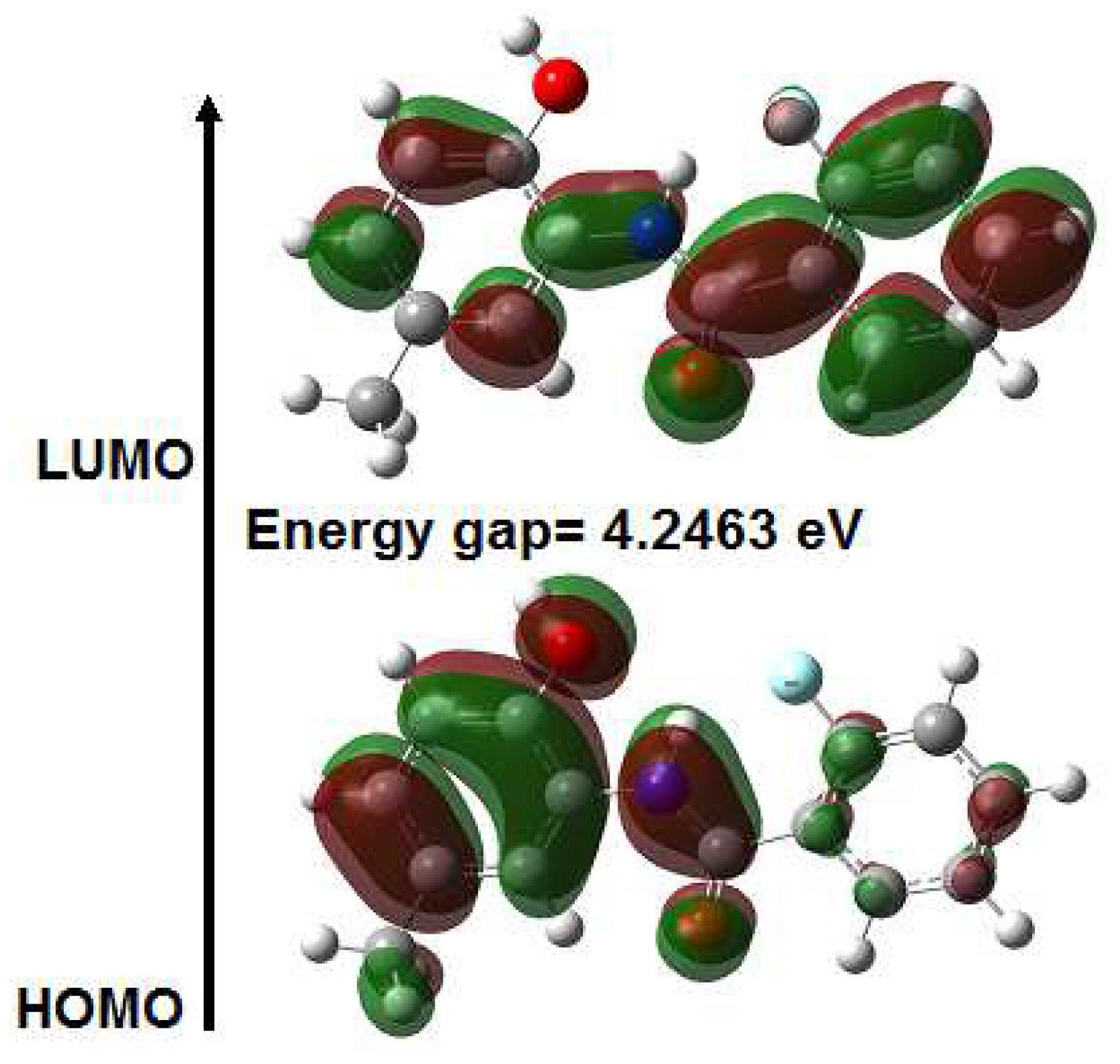
2.4. UV-Vis
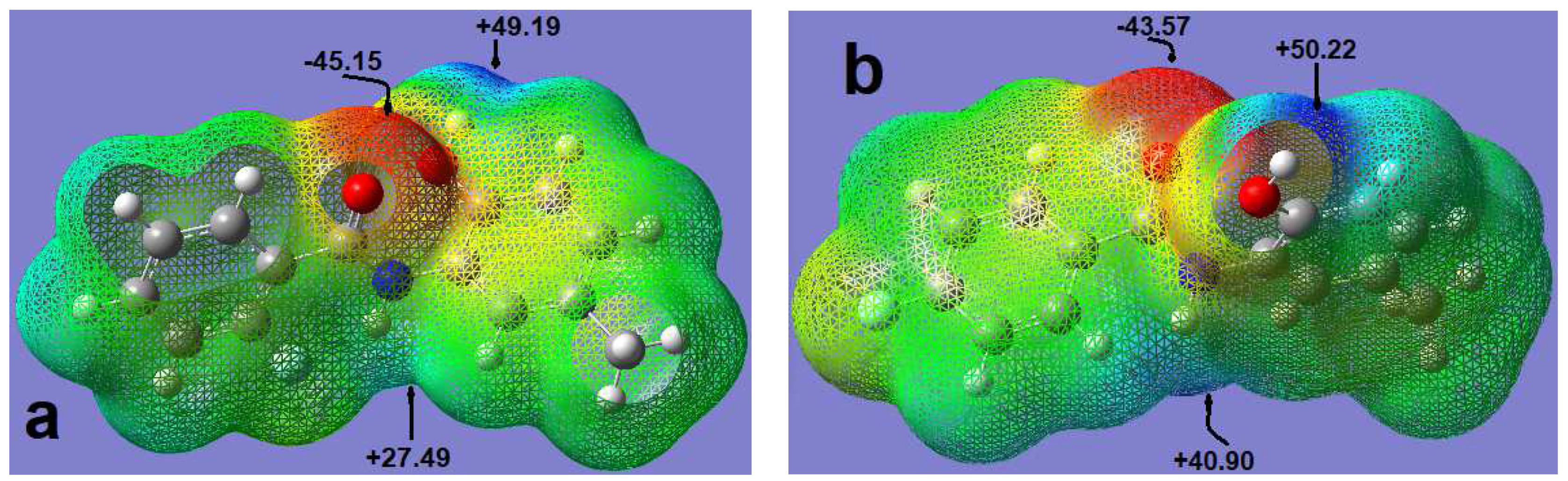
2.5. Molecular Electrostatic Potential (MEP)
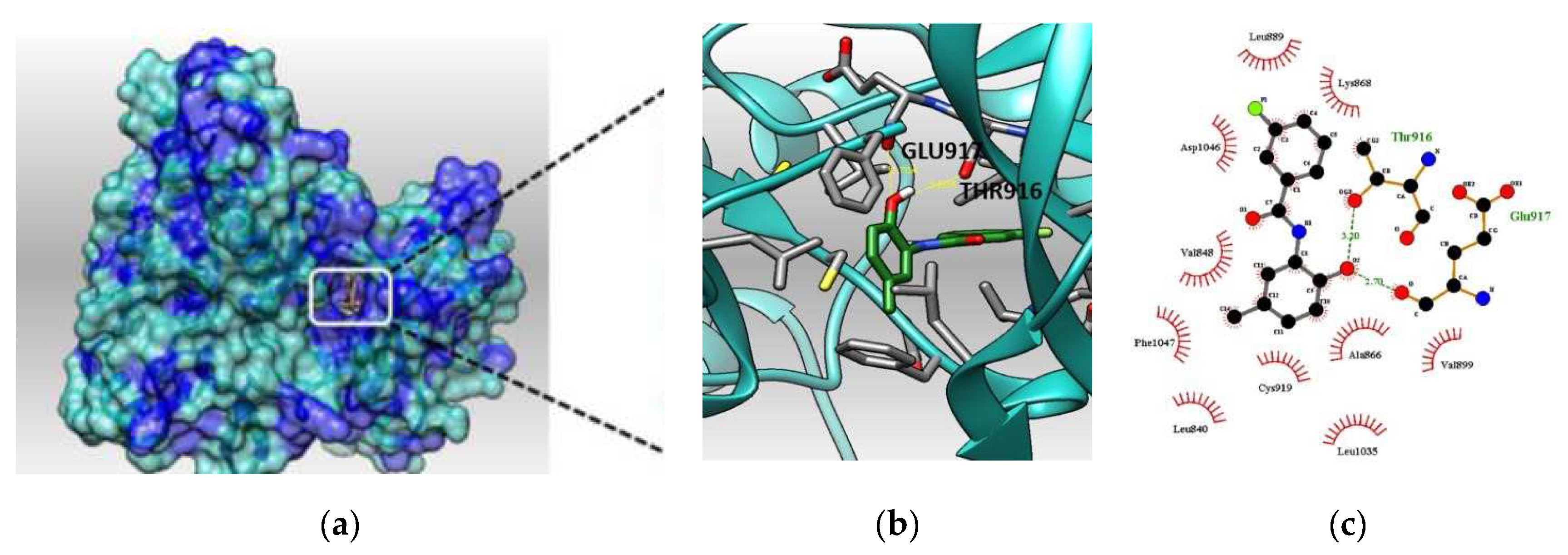
2.6. Docking Studies
3. Materials and Methods
3.1. General Information
3.2. Computational Methods
3.3. Refinement
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ji, X.-Y.; Wang, H.-Q.; Hao, L.-H.; He, W.-Y.; Gao, R.-M.; Li, Y.-P.; Li, Y.-H.; Jiang, J.-D.; Li, Z.-R. Synthesis and antiviral activity of N-phenylbenzamide derivatives, a novel class of enterovirus 71 inhibitors. Molecules 2013, 18, 3630–3640. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Tokunaga, E.; Kobayashi, O.; Hirai, K.; Shibata, N. Current contributions of organofluorine compounds to the agrochemical industry. iScience 2020, 23, 101467. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, D.B.; Karukurichi, K.R.; de la Salud-Bea, R.; Nelson, D.L.; McCune, C.D. Use of fluorinated functionality in enzyme inhibitor development: Mechanistic and analytical advantages. J. Fluor. Chem. 2008, 129, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Filler, R.; Saha, R. Fluorine in medicinal chemistry: A century of progress and a 60-year retrospective of selected highlights. Future Med. Chem. 2009, 1, 777–791. [Google Scholar] [CrossRef] [PubMed]
- Biffinger, J.C.; Kim, H.W.; DiMagno, S.G. The polar hydrophobicity of fluorinated compounds. ChemBioChem 2004, 5, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, D.B.; Bose, M. (α-Monofluoroalkyl)phosphonates: A class of iso-acidic and “tunable” mimics of biological phosphates. J. Fluor. Chem. 2001, 112, 13–33. [Google Scholar] [CrossRef]
- Moreno-Fuquen, R.; Mariño, N.J.; Kennedy, A.R. Crystal structure of N-(2-hydroxy-5-methylphenyl)benzamide. Acta Crystallogr. E: Crystallogr. Commun. 2015, 71, o943. [Google Scholar] [CrossRef]
- Montalbetti, C.A.G.N.; Falque, V. Amide bond formation, and peptide coupling. Tetrahedron 2005, 61, 10827–10852. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Emsley, J. The composition, structure and hydrogen bonding of the β-diketones. In Structure and Bonding; Cardine, C., Ed.; Springer: Berlin/Heidelberg, Germany, 1984; Volume 57, pp. 147–191. [Google Scholar]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. Patterns in hydrogen bonding: Functionality and graph set analysis in crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Dunitz, J.D. Organic fluorine: Odd man out. ChemBioChem 2004, 5, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Büyükgüngör, O.; Odabaşoğlu, M. 2-Fluoroanilinium N-(2-fluorophenyl)oxamate. Acta Crystallogr. E: Crystallogr. Commun. 2008, 64, o808. [Google Scholar] [CrossRef] [PubMed]
- Barbarich, T.J.; Rithner, C.D.; Miller, S.M.; Anderson, O.P.; Strauss, S.H. Significant inter- and intramolecular O−H···FC hydrogen bonding. J. Am. Chem. Soc. 1999, 121, 4280–4281. [Google Scholar] [CrossRef]
- Gowda, B.T.; Foro, S.; Sowmya, B.P.; Fuess, H. 3-Chloro-N-(3-chlorophenyl)benzamide. Acta Crystallogr. E: Crystallogr. Commun. 2008, 64, o949. [Google Scholar] [CrossRef]
- Tan, Z.; Bing, Y.; Fang, S.; Kai, Z.; Yan, Y. 2-Chloro-4-fluoro-N-phenylbenzamide. Acta Crystallogr. E: Crystallogr. Commun. 2009, 65, o1757. [Google Scholar] [CrossRef]
- Fun, H.-K.; Chantrapromma, S.; Sripet, W.; Ruanwas, P.; Boonnak, N. 4-Bromo-N-phenylbenzamide. Acta Crystallogr. E: Crystallogr. Commun. 2012, 68, o1269–o1270. [Google Scholar] [CrossRef]
- Fukui, K.; Yonezawa, T.; Shingu, H.J. A Molecular orbital theory of reactivity in aromatic hydrocarbons. J. Chem. Phys. 1952, 20, 722–725. [Google Scholar] [CrossRef]
- Politzer, P.; Truhlar, D.G. Chemical Applications of Atomic and Molecular Electrostatic Potentials; Plenum Press: New York, NY, USA, 1981; pp. 1–6. [Google Scholar]
- Politzer, P.; Murray, J.S. The fundamental nature and role of the electrostatic potential in atoms and molecules. Theor. Chem. Acc. 2002, 108, 134–149. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision B. 01.; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.J.; Rixe, O. Axitinib—A selective inhibitor of the vascular endothelial growth factor (VEGF) receptor. Target. Oncol. 2009, 4, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Tresaugues, L.; Roos, A.; Arrowsmith, C.H.; Berglund, H.; Bountra, C.; Collins, R.; Edwards, A.M.; Flodin, S.; Flores, A.; Graslund, S.; et al. Crystal structure of VEGFR1 in complex with N-(4-Chlorophenyl)-2-((pyridin-4-ylmethyl)amino)benzamide. RCSB PDB 2009. [Google Scholar] [CrossRef]
- Norman, M.H.; Liu, L.; Lee, M.; Xi, N.; Fellows, I.; D’Angelo, N.D.; Dominguez, C.; Rex, K.; Bellon, S.F.; Kim, T.S.; et al. Structure-based design of novel class II c-Met inhibitors: Identification of pyrazolone-based derivatives. J. Med. Chem. 2012, 55, 1858–1867. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 31, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. 1988, A38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. 1988, B37, 785–789. [Google Scholar] [CrossRef]
- Nielsen, A.B.; Holder, A.J. Gauss View 5.0, User’s Reference; Gaussian Inc.: Pittsburgh, PA, USA, 2009. [Google Scholar]
- Jamroz, M.H. Vibrational energy distribution analysis (VEDA): Scopes and limitations. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 114, 220–230. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isomer | D-H···A | D-H | H···A | D···A | D-H···A |
|---|---|---|---|---|---|
| O2-H02∙∙∙O1 i | 0.87(3) | 1.78(3) | 2.6402(16) | 170(3) | |
| C6-H6∙∙∙O2 ii | 0.93 | 2.52 | 3.394(2) | 156.2 | |
| o-FPhB | C5-H5∙∙∙F1 ii | 0.93 | 2.96 | 3.632(2) | 130.7 |
| C2-F1∙∙∙Cg1 | 3.6441(13) | ||||
| N1-H1∙∙∙F1 | 0.86 | 1.99 | 2.6974(15) | 138.6 | |
| O2-H02∙∙∙O1 iii | 0.80(3) | 1.85(3) | 2.6454(18) | 175(3) | |
| m-FPhB | C6-H6∙∙∙O2 iv | 0.93 | 2.52 | 3.376(2) | 152.9 |
| C14-H141∙∙∙F1 v | 0.96 | 2.67 | 3.385(3) | 131.4 | |
| C7-O1···Cg1 | 3.8939(17) | ||||
| C5-F1∙∙∙Cg2 | 3.583(2) | ||||
| N1-H1∙∙∙O1 vi | 0.86 | 2.01 | 2.807(3) | 153.9 | |
| p-FPhB | C5-H5∙∙∙F1 vii | 0.93 | 2.58 | 3.499(5) | 169.6 |
| O2-HO2∙∙∙O1 viii | 0.87(5) | 1.87(3) | 2.663(3) | 152(4) | |
| C2-H2∙∙∙O2 ix | 0.93 | 2.58 | 3.499(4) | 169.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno-Fuquen, R.; Mariño-Ocampo, N.; Tenorio, J.C.; Ellena, J.; Kennedy, A.R. Activation–Deactivation of Inter-Peptide Bond in Fluoro-N-(2-hydroxy-5-methyl phenyl)benzamide Isomers, Induced by the Position of the Halogen Atom in the Benzene Ring. Molbank 2022, 2022, M1416. https://doi.org/10.3390/M1416
Moreno-Fuquen R, Mariño-Ocampo N, Tenorio JC, Ellena J, Kennedy AR. Activation–Deactivation of Inter-Peptide Bond in Fluoro-N-(2-hydroxy-5-methyl phenyl)benzamide Isomers, Induced by the Position of the Halogen Atom in the Benzene Ring. Molbank. 2022; 2022(3):M1416. https://doi.org/10.3390/M1416
Chicago/Turabian StyleMoreno-Fuquen, Rodolfo, Nory Mariño-Ocampo, Juan Carlos Tenorio, Javier Ellena, and Alan R. Kennedy. 2022. "Activation–Deactivation of Inter-Peptide Bond in Fluoro-N-(2-hydroxy-5-methyl phenyl)benzamide Isomers, Induced by the Position of the Halogen Atom in the Benzene Ring" Molbank 2022, no. 3: M1416. https://doi.org/10.3390/M1416
APA StyleMoreno-Fuquen, R., Mariño-Ocampo, N., Tenorio, J. C., Ellena, J., & Kennedy, A. R. (2022). Activation–Deactivation of Inter-Peptide Bond in Fluoro-N-(2-hydroxy-5-methyl phenyl)benzamide Isomers, Induced by the Position of the Halogen Atom in the Benzene Ring. Molbank, 2022(3), M1416. https://doi.org/10.3390/M1416