
Design, Synthesis and 5-HT1A Binding Affinity of N-(3-(4-(2-Methoxyphenyl)piperazin-1-yl)propyl)tricyclo[3.3.1.13,7]decan-1-amine and N-(3-(4-(2-Methoxyphenyl)piperazin-1-yl)propyl)-3,5-dimethyl-tricylo[3.3.1.13,7]decan-1-amine



Abstract
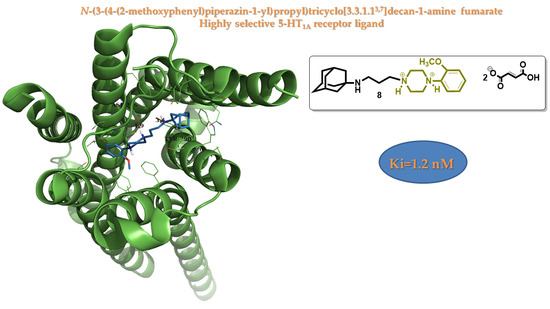
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
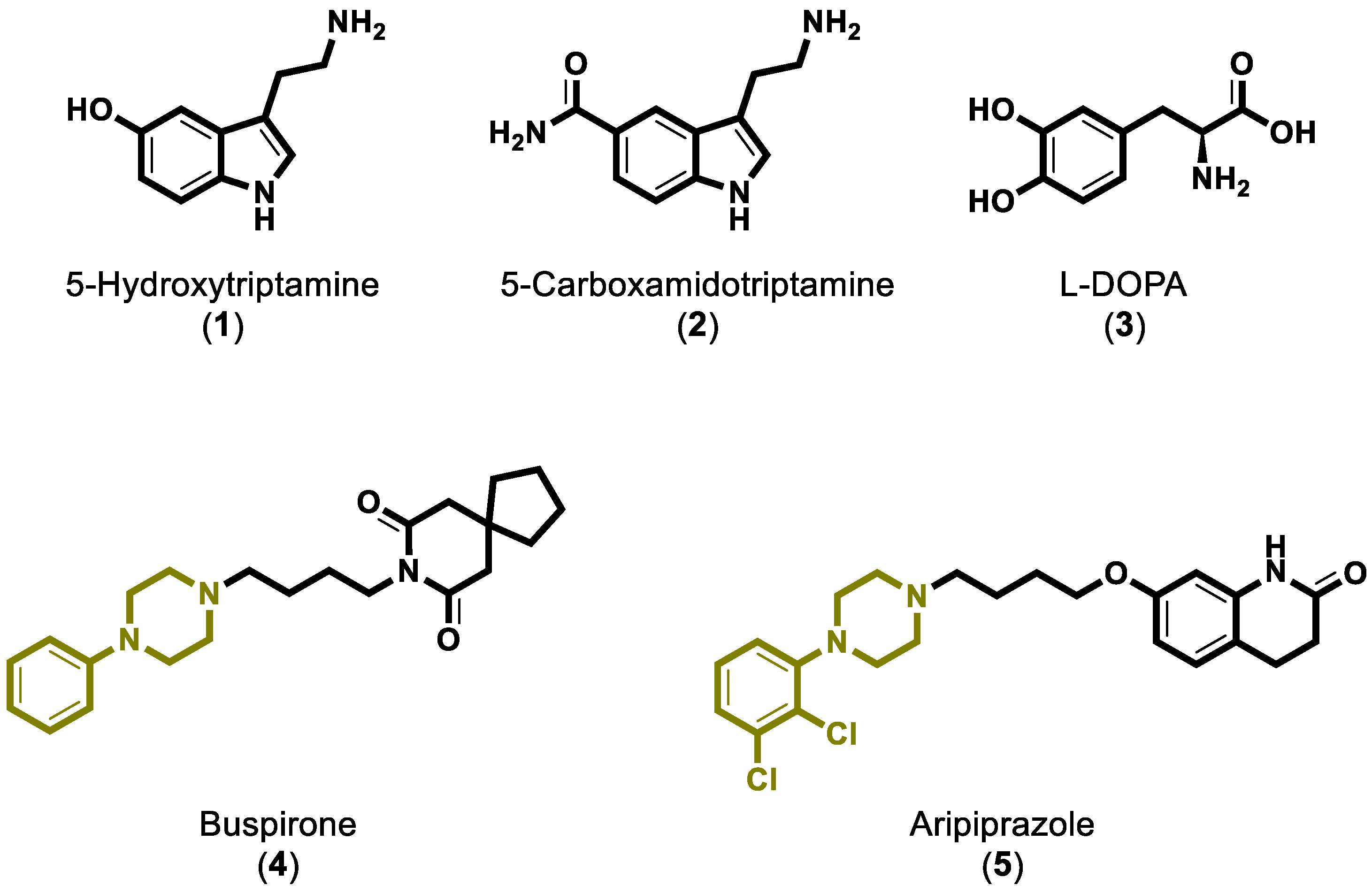
1. Introduction
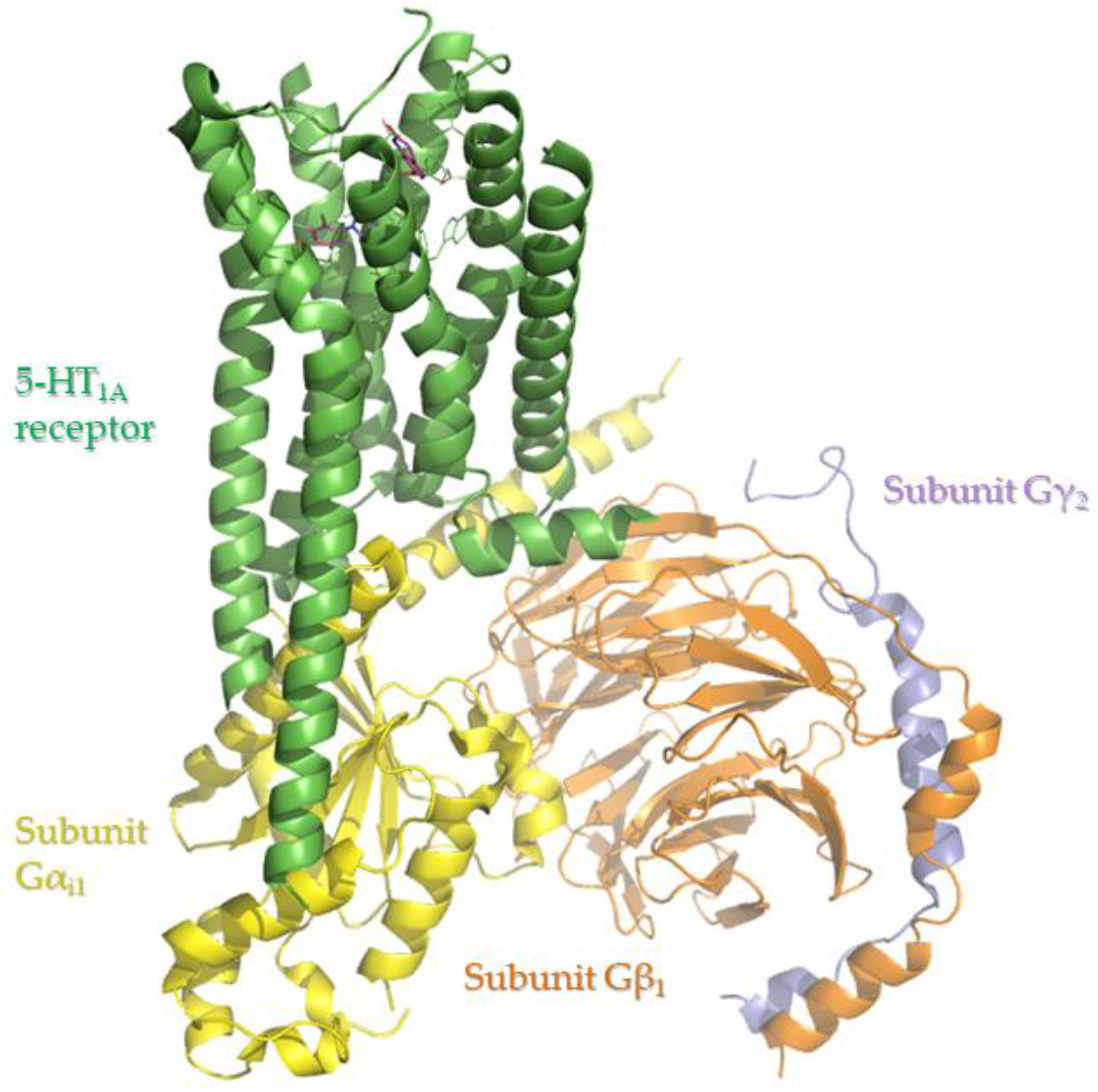
2. Results and Discussion
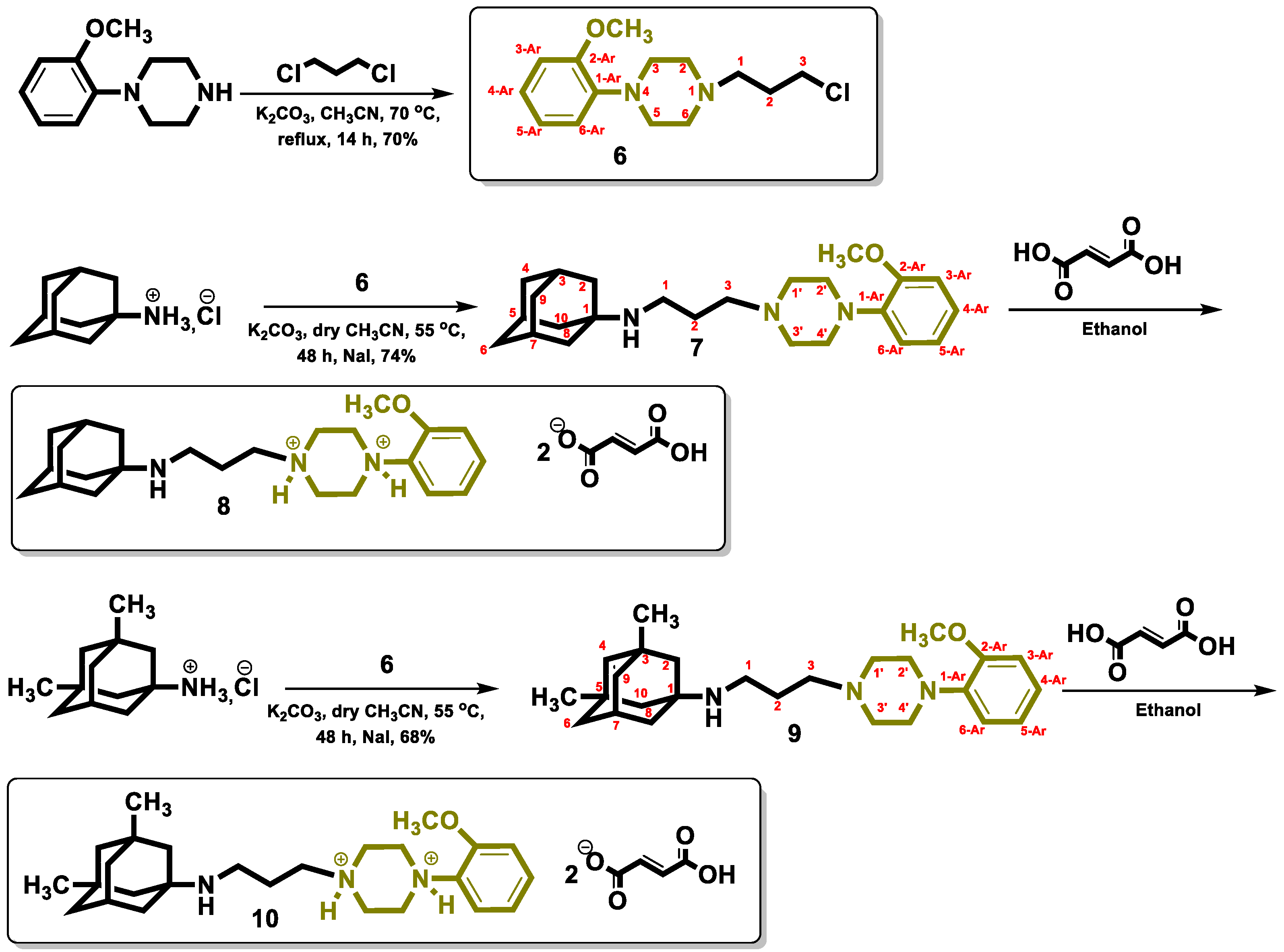
2.1. Chemistry
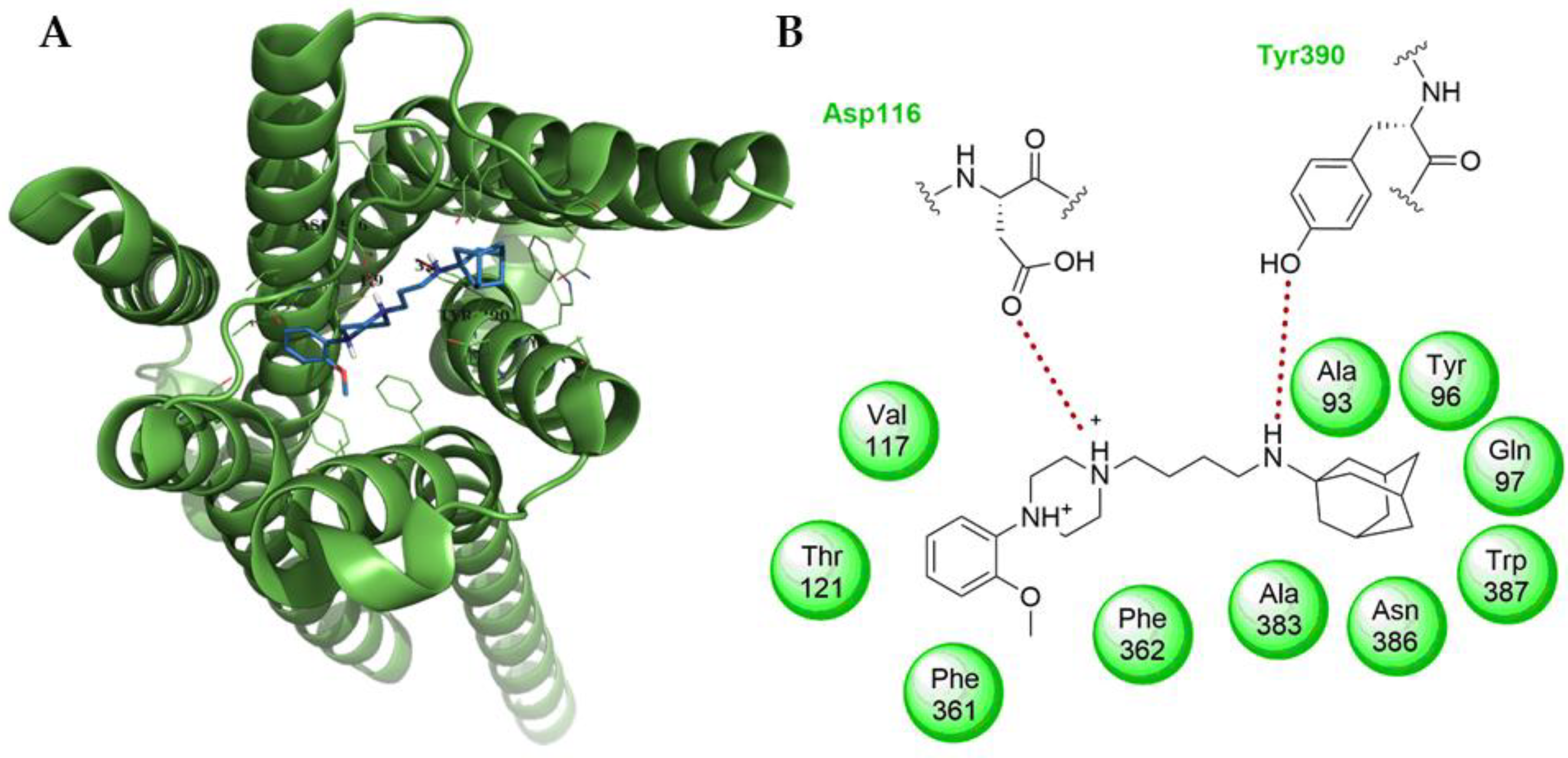
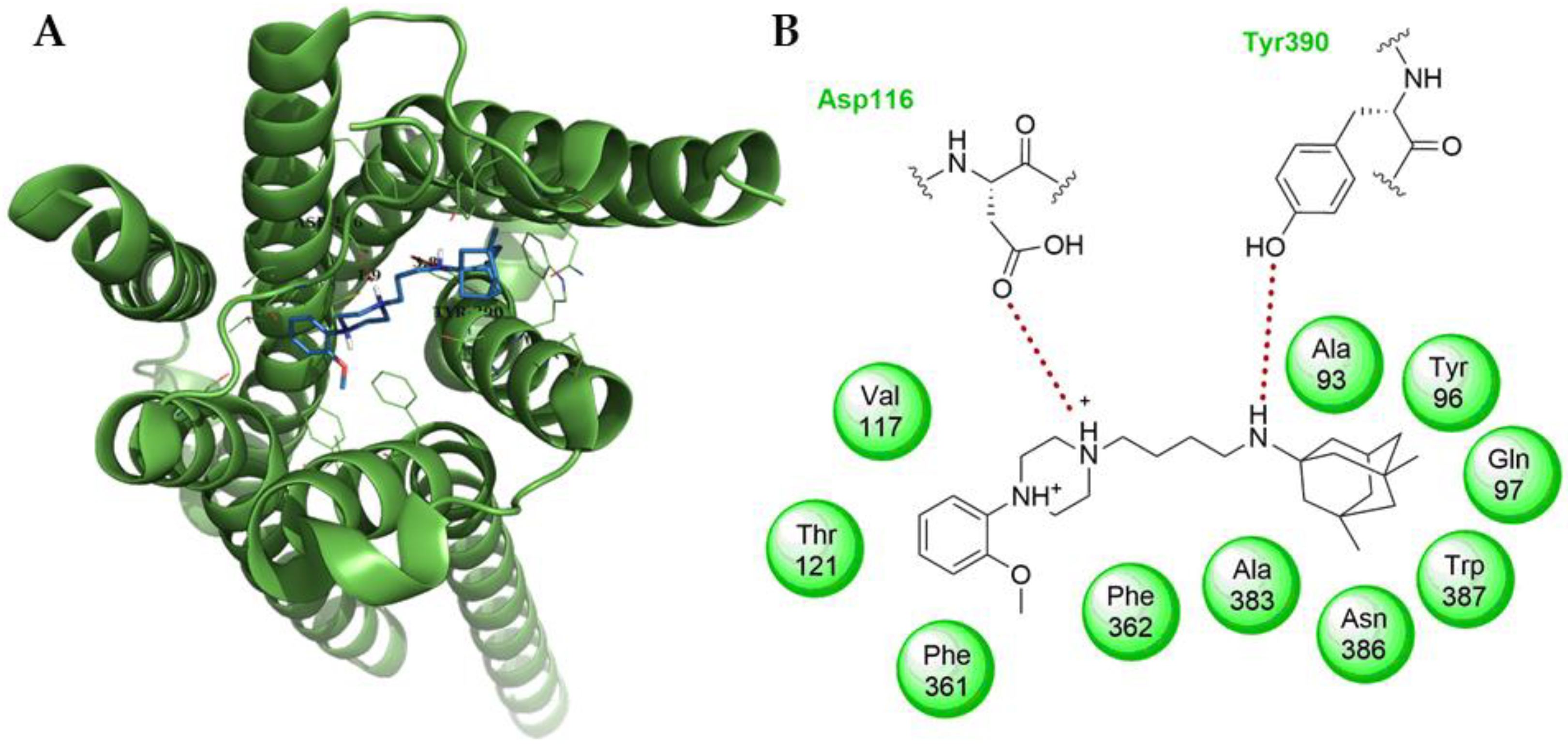
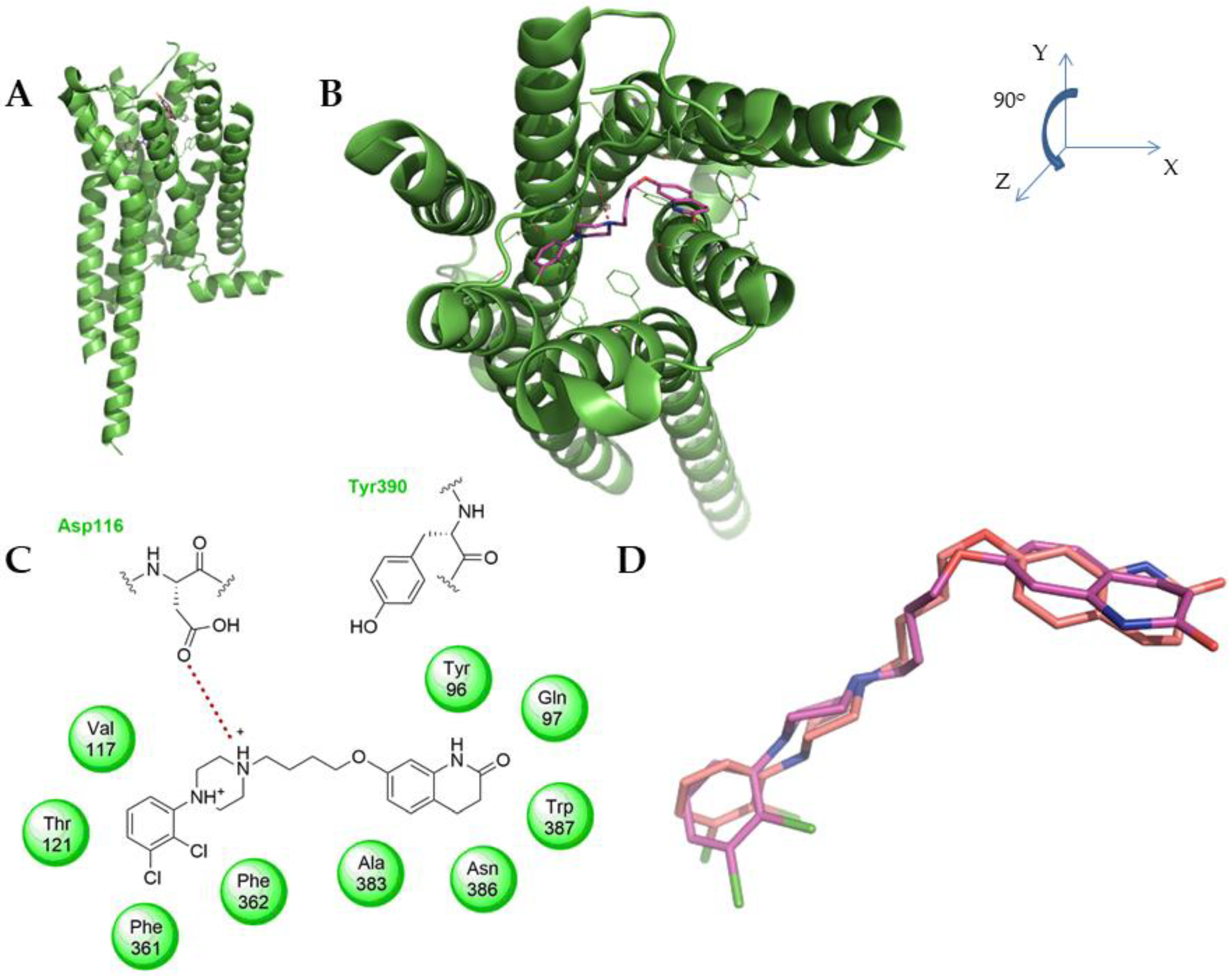
2.2. Biological Activity
3. Materials and Methods
3.1. Chemistry
3.2. Synthesis
3.3. Computational
3.4. Ligand and Protein Preparation
3.5. Model Confidence Experiment
3.6. Biology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schioth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Huang, Y.; Todd, N.; Thathiah, A. The role of GPCRs in neurodegenerative diseases: Avenues for therapeutic intervention. Curr. Opin. Pharmacol. 2017, 32, 96–110. [Google Scholar] [CrossRef]
- Polter, A.M.; Li, X. 5-HT1A receptor-regulated signal transduction pathways in brain. Cell. Signal. 2010, 22, 1406–1412. [Google Scholar] [CrossRef] [Green Version]
- Staron, J.; Bugno, R.; Hogendorf, A.S.; Bojarski, A.J. 5-HT1A receptor ligands and their therapeutic applications: Review of new patents. Expert Opin. Ther. Pat. 2018, 28, 679–689. [Google Scholar] [CrossRef]
- Albert, P.R.; Vahid-Ansari, F. The 5-HT1A receptor: Signaling to behavior. Biochimie 2019, 161, 34–45. [Google Scholar] [CrossRef]
- Popova, N.K.; Naumenko, V.S. 5-HT1A receptor as a key player in the brain 5-HT system. Rev. Neurosci. 2013, 24, 191–204. [Google Scholar] [CrossRef]
- Xu, P.; Huang, S.; Zhang, H.; Mao, C.; Zhou, X.E.; Cheng, X.; Simon, I.A.; Shen, D.D.; Yen, H.Y.; Robinson, C.V.; et al. Structural insights into the lipid and ligand regulation of serotonin receptors. Nature 2021, 592, 469–473. [Google Scholar] [CrossRef]
- Perrone, R.; Berardi, F.; Colabufo, N.A.; Leopoldo, M.; Tortorella, V.; Fiorentini, F.; Olgiati, V.; Ghiglieri, A.; Govoni, S. High affinity and selectivity on 5-HT1A receptor of 1-aryl-4-[1-tetralin)alkyl]piperazines. 2. J. Med. Chem. 1995, 38, 942–949. [Google Scholar] [CrossRef]
- Napoliello, M.J.; Domantay, A.G. Buspirone: A Worldwide Update. Br. J. Psychiatry 2018, 159, 40–44. [Google Scholar] [CrossRef]
- Di Sciascio, G.; Riva, M.A. Aripiprazole: From pharmacological profile to clinical use. Neuropsychiatr. Dis. Treat. 2015, 11, 2635–2647. [Google Scholar] [CrossRef] [Green Version]
- Giannakopoulou, E.; Pardali, V.; Skrettas, I.; Zoidis, G. Transesterification instead of N-Alkylation: An Intriguing Reaction. ChemistrySelect 2019, 4, 3195–3198. [Google Scholar] [CrossRef]
- Kolocouris, N.; Zoidis, G.; Fytas, C. Facile synthetic routes to 2-oxo-1-adamantanalkanoic acids. Synlett 2007, 2007, 1063–1066. [Google Scholar] [CrossRef]
- Zoidis, G.; Benaki, D.; Myrianthopoulos, V.; Naesens, L.; De Clercq, E.; Mikros, E.; Kolocouris, N. Synthesis of 1, 2-annulated adamantane heterocycles: Structural determination studies of a bioactive cyclic sulfite. Tetrahedron Lett. 2009, 50, 2671–2675. [Google Scholar] [CrossRef]
- Tsatsaroni, A.; Zoidis, G.; Zoumpoulakis, P.; Tsotinis, A.; Taylor, M.C.; Kelly, J.M.; Fytas, G. An E/Z conformational behaviour study on the trypanocidal action of lipophilic spiro carbocyclic 2,6-diketopiperazine-1-acetohydroxamic acids. Tetrahedron Lett. 2013, 54, 3238–3240. [Google Scholar] [CrossRef] [Green Version]
- Fytas, C.; Zoidis, G.; Fytas, G. A facile and effective synthesis of lipophilic 2, 6-diketopiperazine analogues. Tetrahedron 2008, 64, 6749–6754. [Google Scholar] [CrossRef]
- Hamon, M. The main features of central 5-HT 1A receptors. In Serotoninergic Neurons and 5-HT Receptors in the CNS; Springer: Berlin/Heidelberg, Germany, 2000; pp. 239–268. [Google Scholar]
- Hamon, M.; Lanfumey, L.; El Mestikawy, S.; Boni, C.; Miquel, M.-C.; Bolanos, F.; Schechter, L.; Gozlan, H. The main features of central 5-HT1 receptors. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 1990, 3, 349–360. [Google Scholar]
- McGann, M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef]
- McGaughey, G.B.; Sheridan, R.P.; Bayly, C.I.; Culberson, J.C.; Kreatsoulas, C.; Lindsley, S.; Maiorov, V.; Truchon, J.F.; Cornell, W.D. Comparison of topological, shape, and docking methods in virtual screening. J. Chem. Inf. Modeling 2007, 47, 1504–1519. [Google Scholar] [CrossRef]
- McGann, M.R.; Almond, H.R.; Nicholls, A.; Grant, J.A.; Brown, F.K. Gaussian docking functions. Biopolymers 2003, 68, 76–90. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, D.A.; Renock, S.; Arrington, E.; Chiodo, L.A.; Liu, L.X.; Sibley, D.R.; Roth, B.L.; Mailman, R. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology 2003, 28, 1400–1411. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, D.; Caballero, J. Is It Reliable to Take the Molecular Docking Top Scoring Position as the Best Solution without Considering Available Structural Data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef] [Green Version]
- Bell, E.W.; Zhang, Y. DockRMSD: An open-source tool for atom mapping and RMSD calculation of symmetric molecules through graph isomorphism. J. Cheminform. 2019, 11, 40. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, P.C.; Nicholls, A. Conformer generation with OMEGA: Learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [Google Scholar] [CrossRef]
- Hawkins, P.C.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System; Version 1.4.1; Schrodinger, LLC: New York, NY, USA.
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Kelley, B.P.; Brown, S.P.; Warren, G.L.; Muchmore, S.W. POSIT: Flexible Shape-Guided Docking for Pose Prediction. J. Chem. Inf. Model. 2015, 55, 1771–1780. [Google Scholar] [CrossRef]
- McGann, M. FRED and HYBRID docking performance on standardized datasets. J. Comput.-Aided Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef]
- Cheng, Y.C.; Prusoff, W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzyme reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zoidis, G.; Loza, M.I.; Catto, M. Design, Synthesis and 5-HT1A Binding Affinity of N-(3-(4-(2-Methoxyphenyl)piperazin-1-yl)propyl)tricyclo[3.3.1.13,7]decan-1-amine and N-(3-(4-(2-Methoxyphenyl)piperazin-1-yl)propyl)-3,5-dimethyl-tricylo[3.3.1.13,7]decan-1-amine. Molbank 2022, 2022, M1353. https://doi.org/10.3390/M1353
Zoidis G, Loza MI, Catto M. Design, Synthesis and 5-HT1A Binding Affinity of N-(3-(4-(2-Methoxyphenyl)piperazin-1-yl)propyl)tricyclo[3.3.1.13,7]decan-1-amine and N-(3-(4-(2-Methoxyphenyl)piperazin-1-yl)propyl)-3,5-dimethyl-tricylo[3.3.1.13,7]decan-1-amine. Molbank. 2022; 2022(1):M1353. https://doi.org/10.3390/M1353
Chicago/Turabian StyleZoidis, Grigoris, María Isabel Loza, and Marco Catto. 2022. "Design, Synthesis and 5-HT1A Binding Affinity of N-(3-(4-(2-Methoxyphenyl)piperazin-1-yl)propyl)tricyclo[3.3.1.13,7]decan-1-amine and N-(3-(4-(2-Methoxyphenyl)piperazin-1-yl)propyl)-3,5-dimethyl-tricylo[3.3.1.13,7]decan-1-amine" Molbank 2022, no. 1: M1353. https://doi.org/10.3390/M1353
APA StyleZoidis, G., Loza, M. I., & Catto, M. (2022). Design, Synthesis and 5-HT1A Binding Affinity of N-(3-(4-(2-Methoxyphenyl)piperazin-1-yl)propyl)tricyclo[3.3.1.13,7]decan-1-amine and N-(3-(4-(2-Methoxyphenyl)piperazin-1-yl)propyl)-3,5-dimethyl-tricylo[3.3.1.13,7]decan-1-amine. Molbank, 2022(1), M1353. https://doi.org/10.3390/M1353