
In Silico Evaluation of a Promising Key Intermediate Thieno [2,3-d] Pyrimidine Derivative with Expected JAK2 Kinase Inhibitory Activity


Abstract
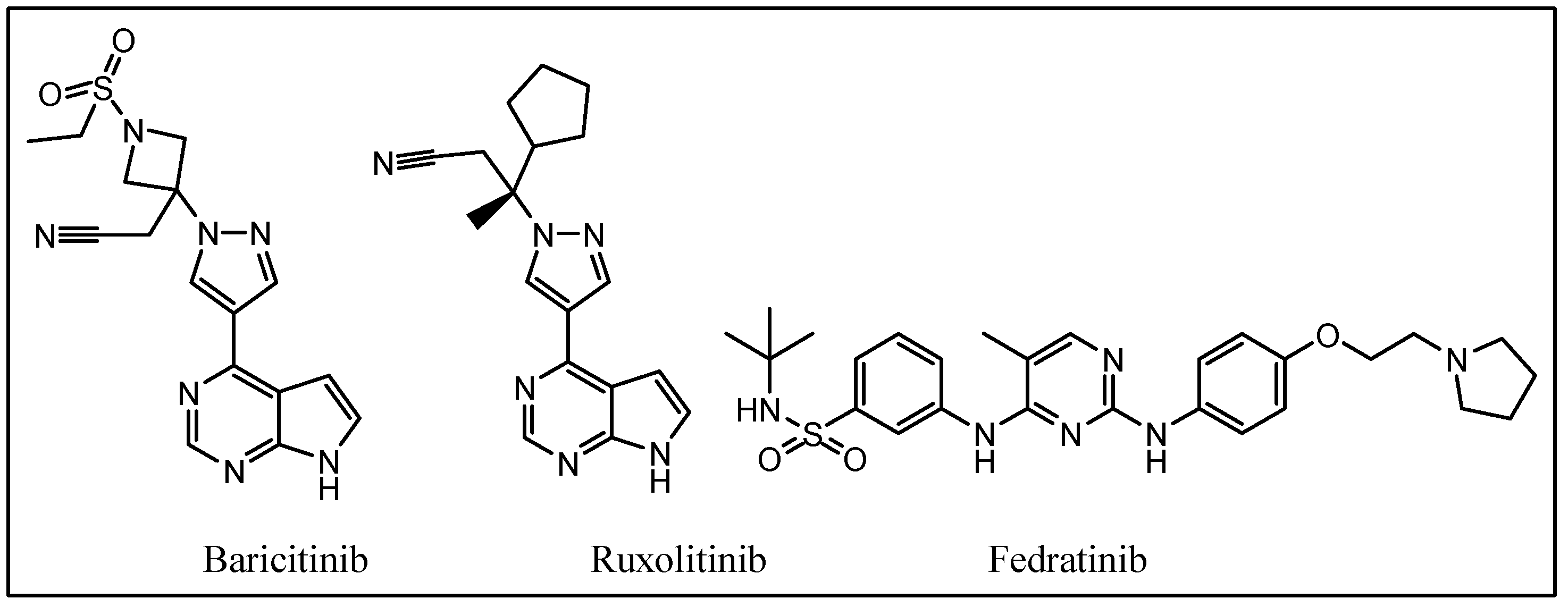
:1. Introduction
2. Results and Discussion
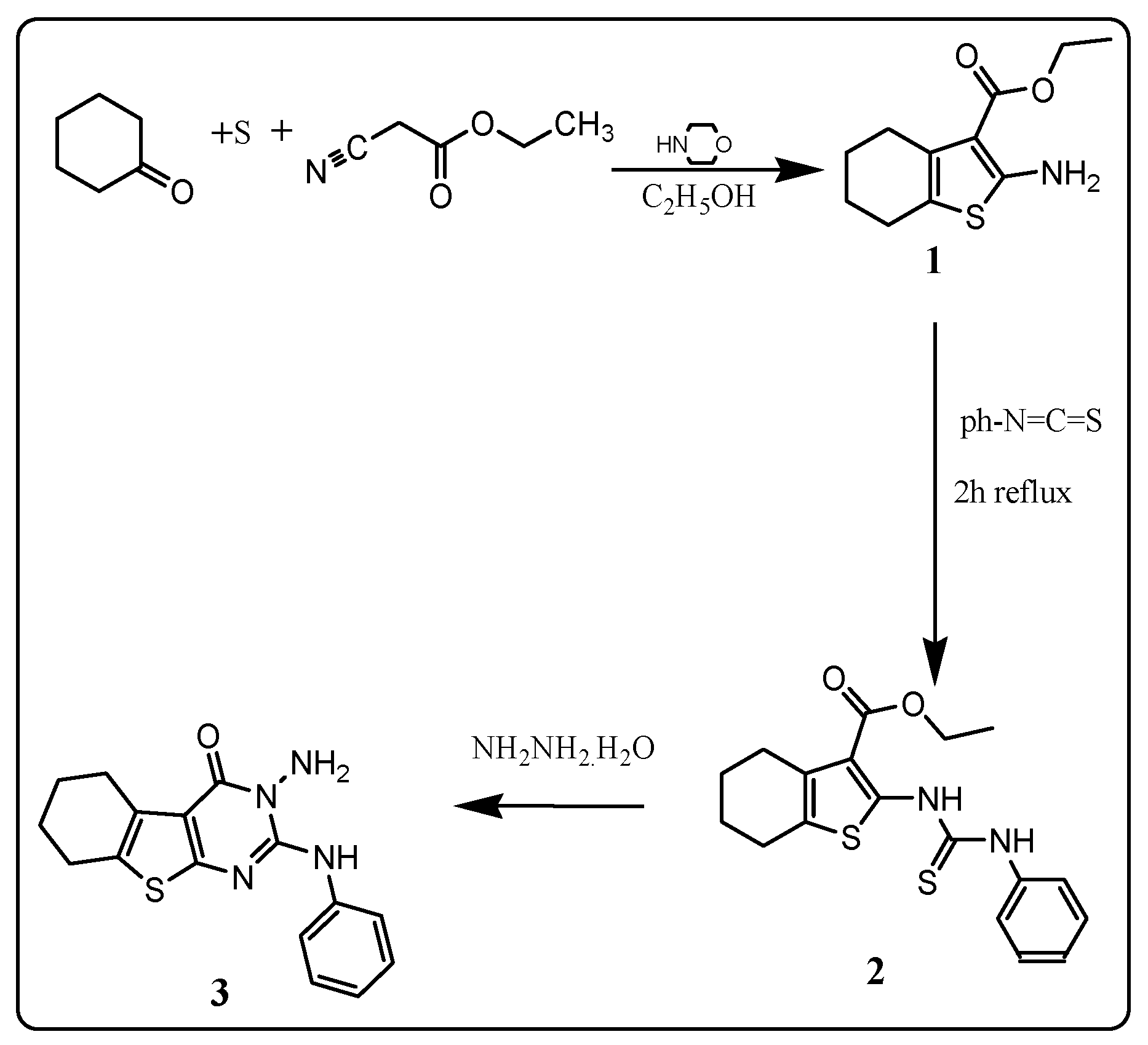
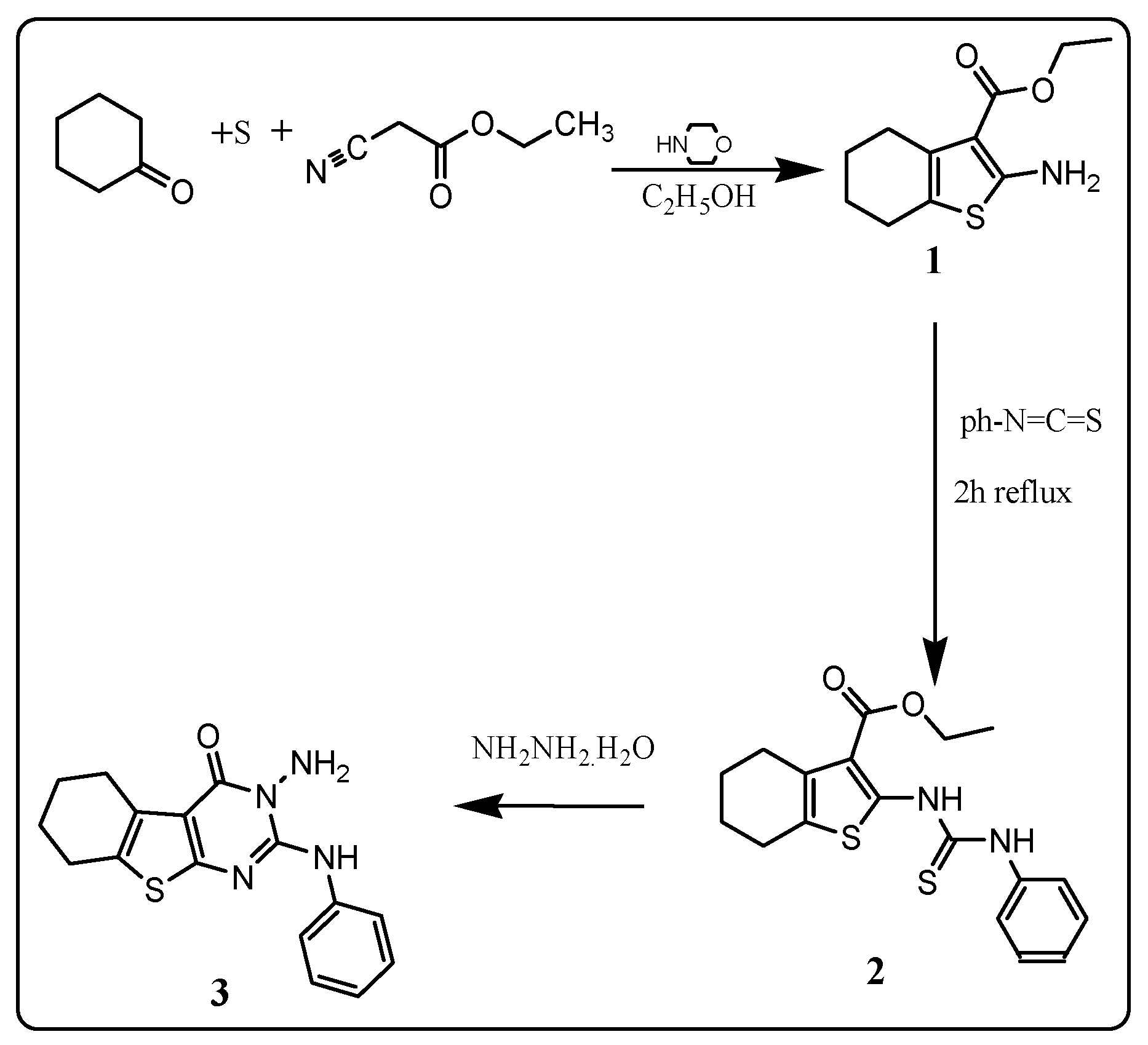
2.1. Chemistry
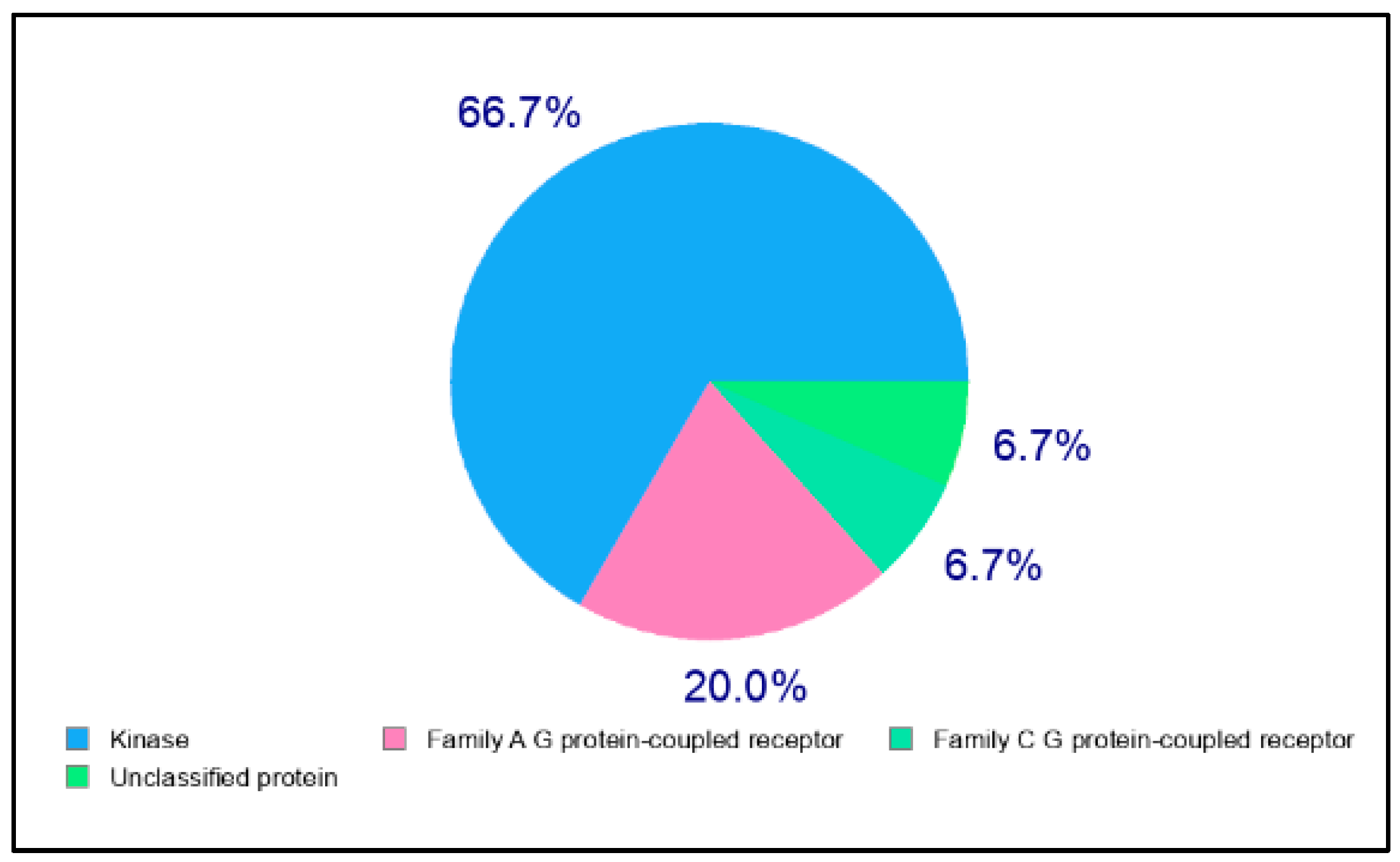
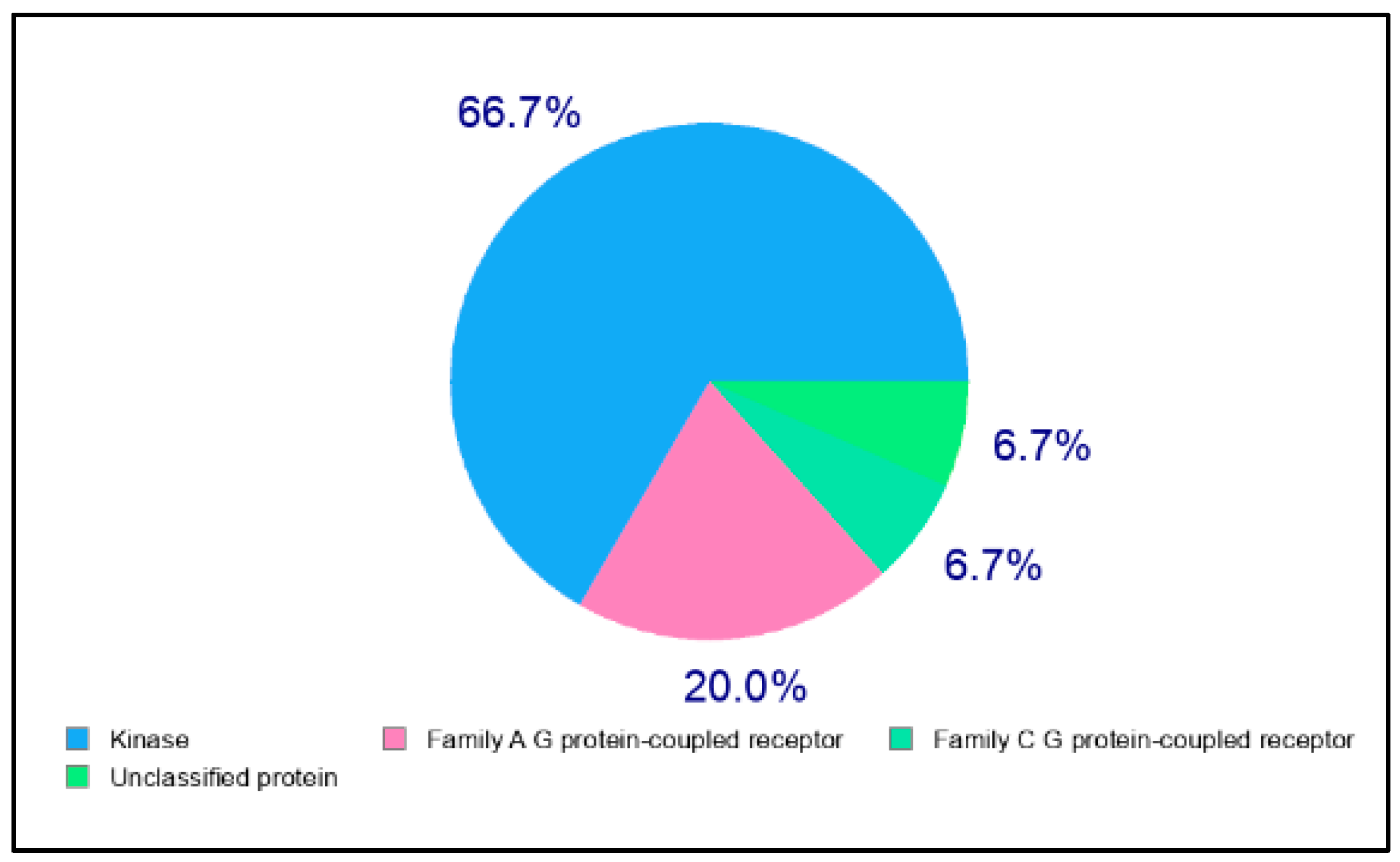
2.2. In Silico Target Prediction
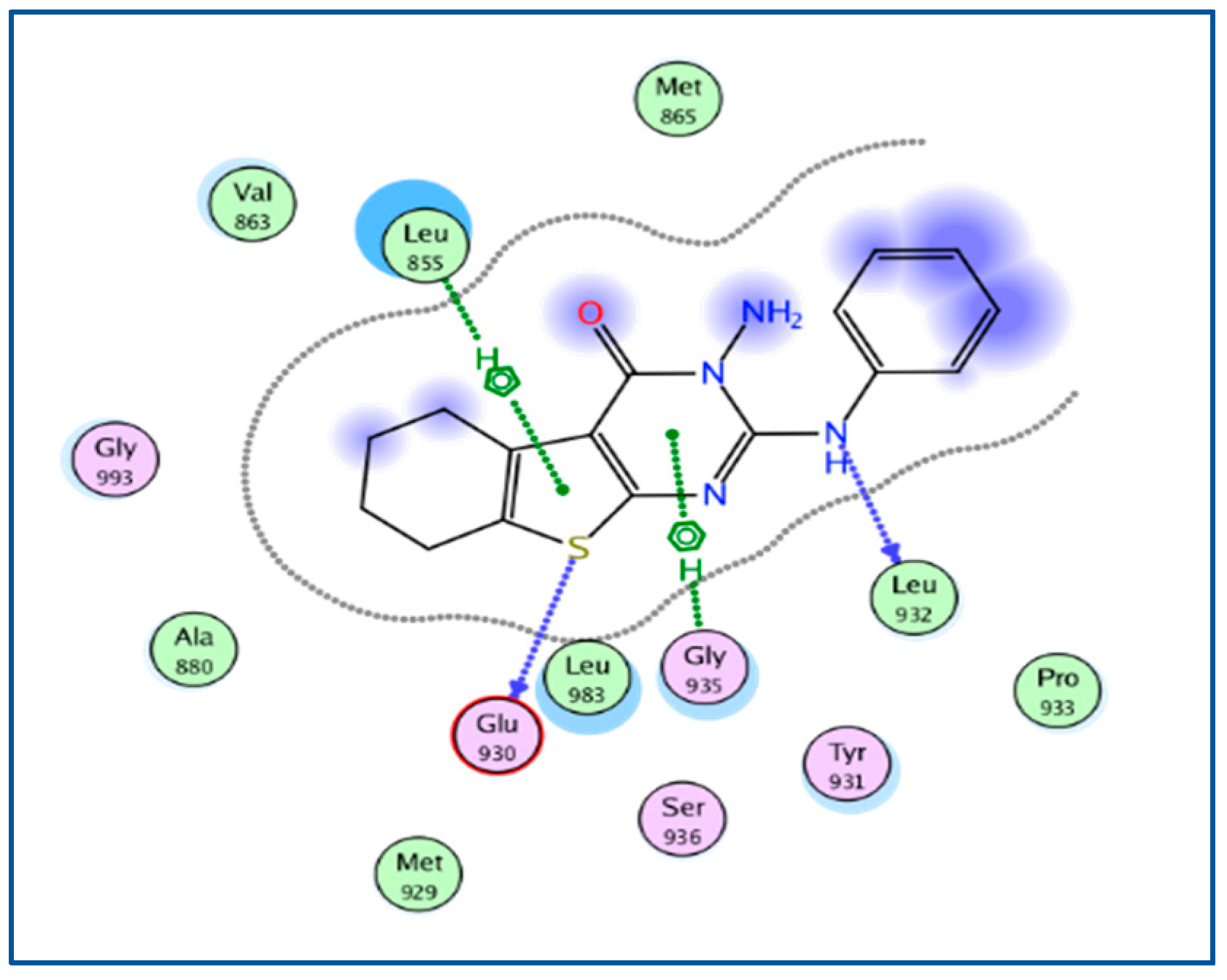
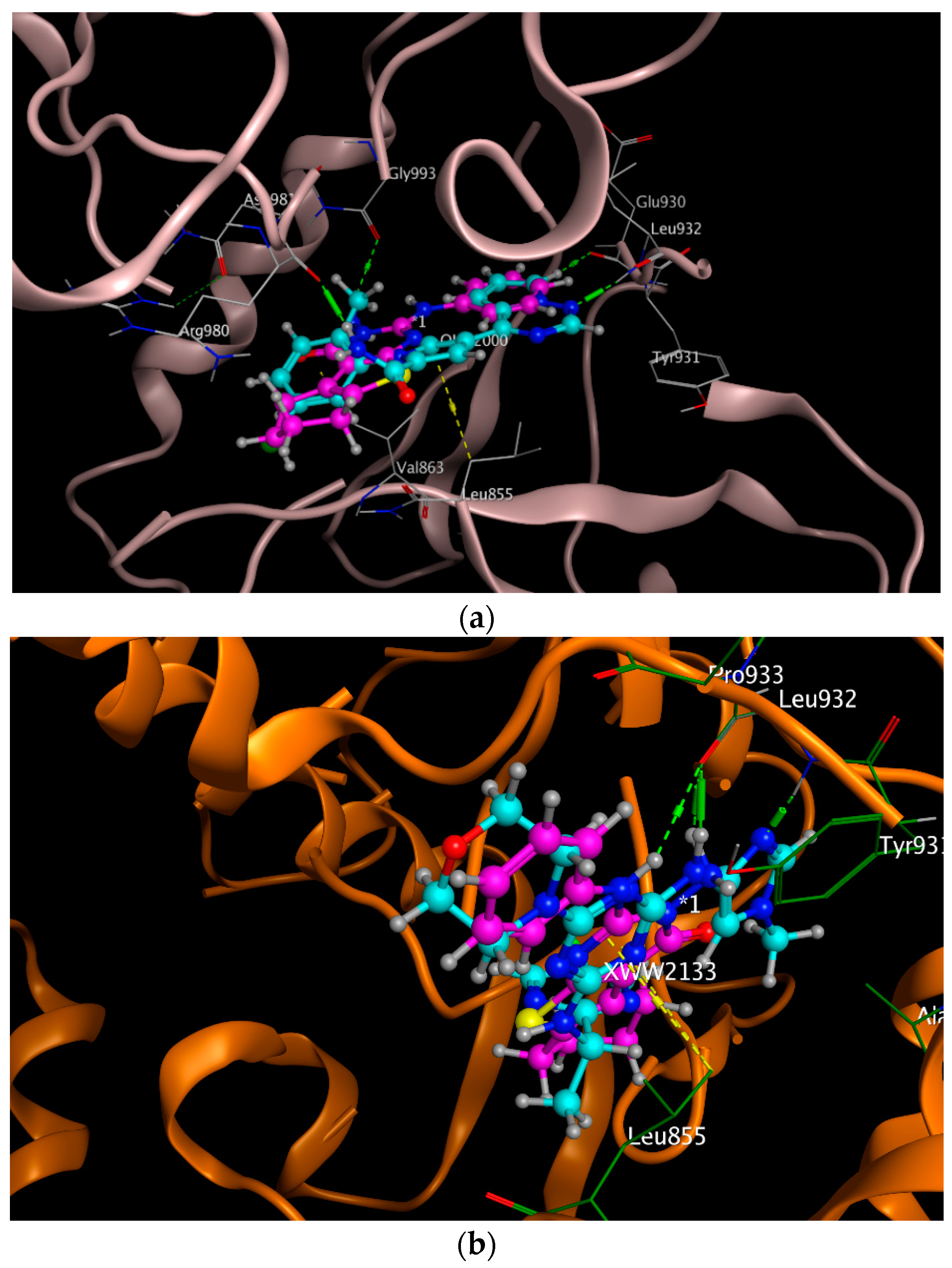
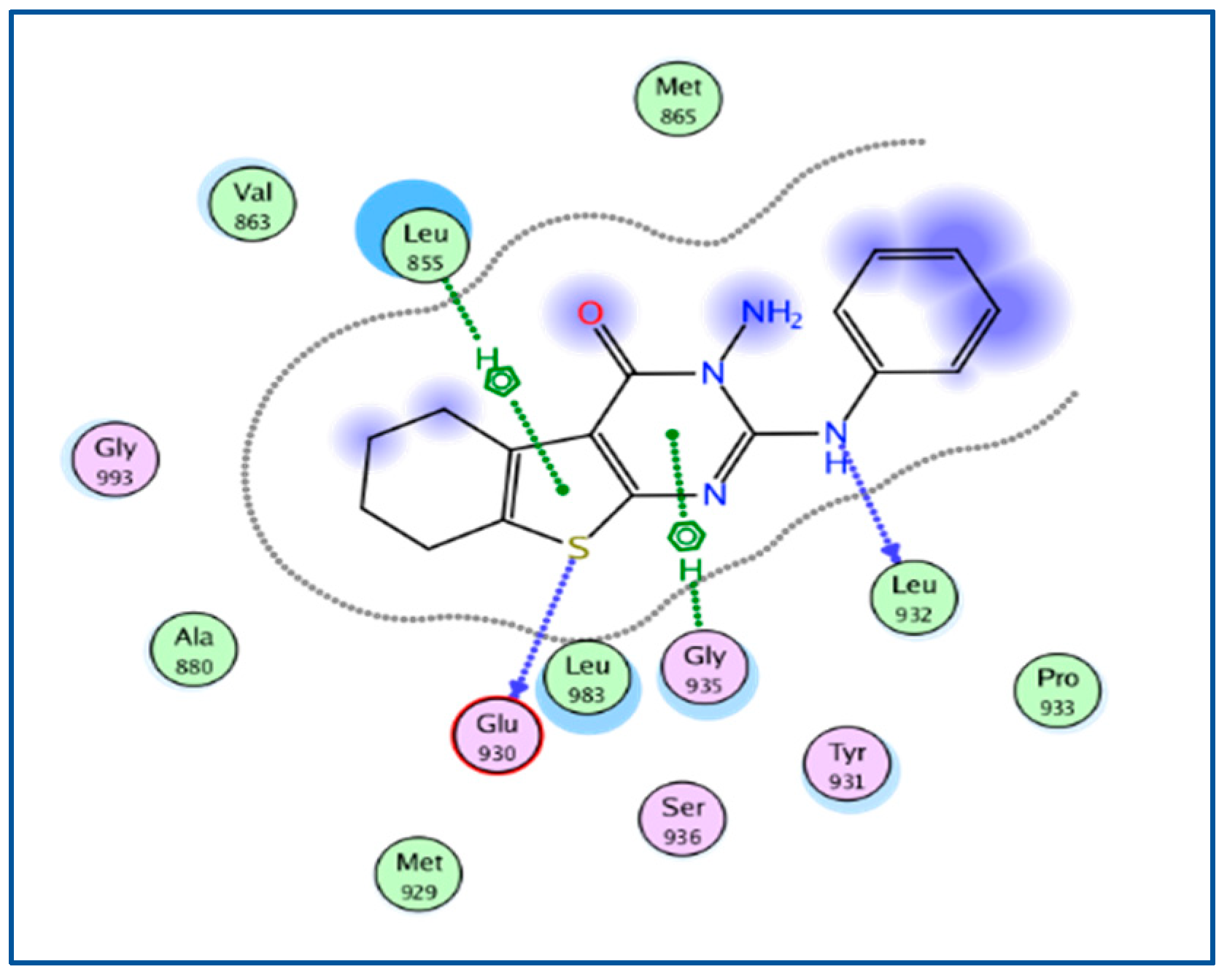
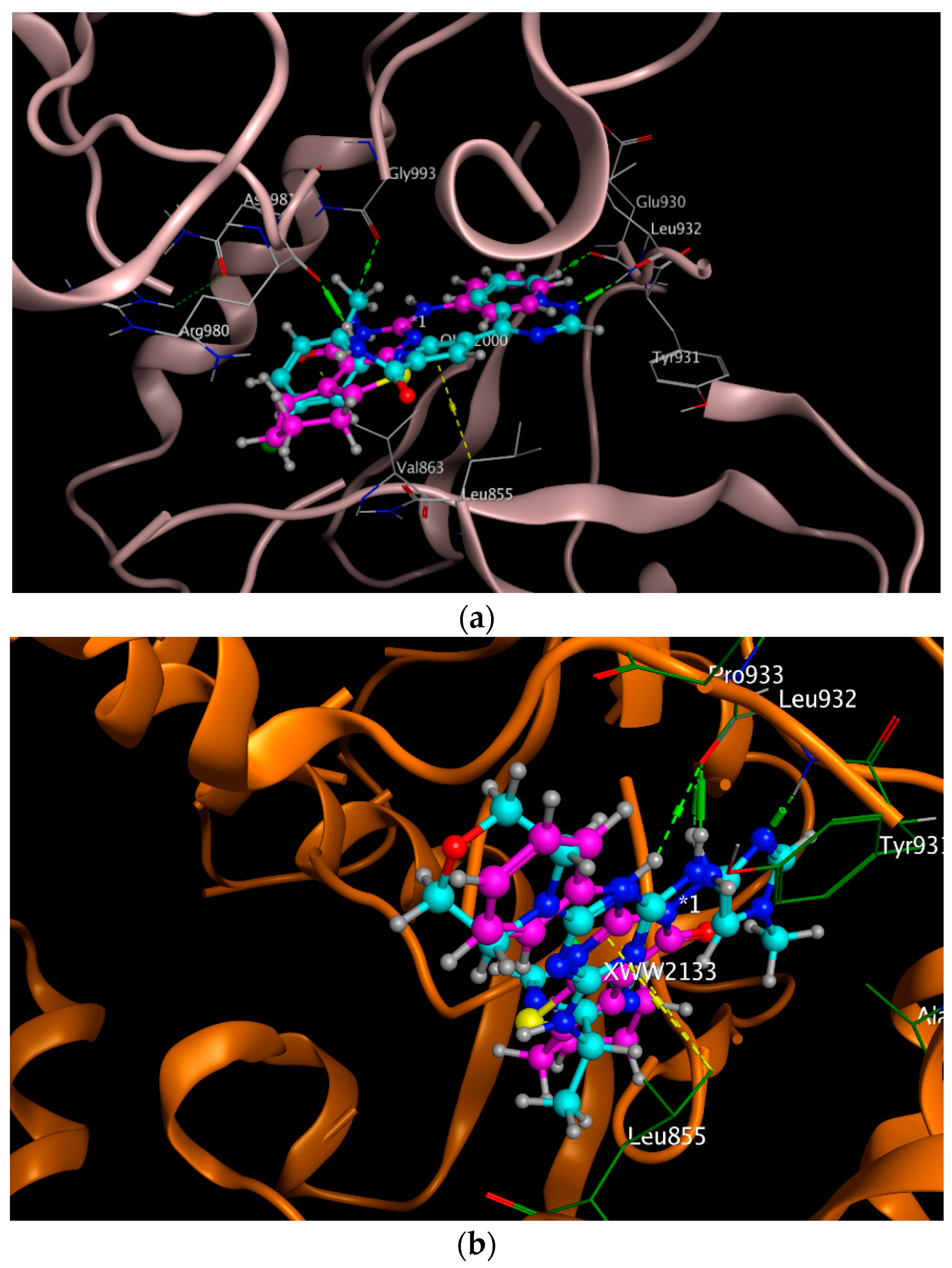
2.3. Molecular Docking
2.4. In Vitro Kinase Screening
2.5. In Vitro Cytotoxic Evaluation against Three Cancerous Cell Lines
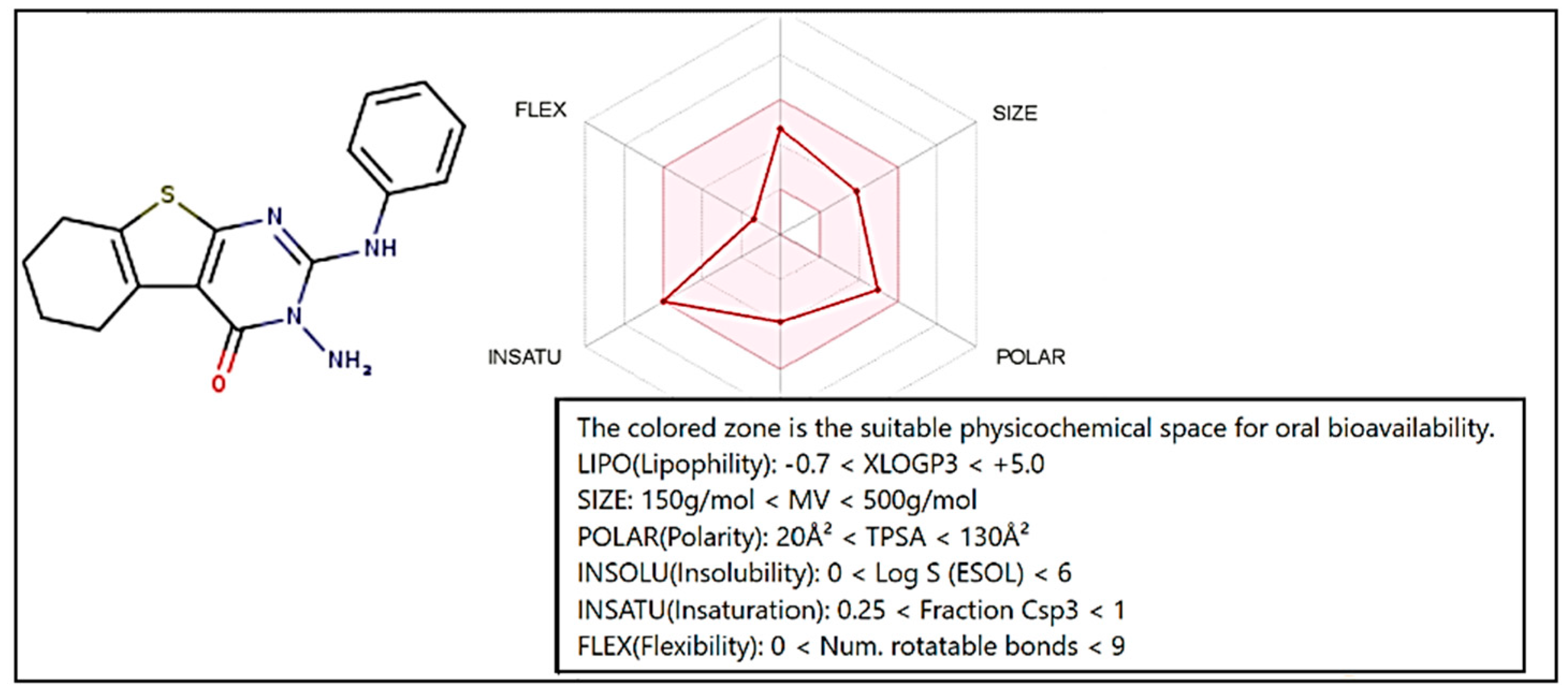
2.6. Pharmacokinetics Study
3. Materials and Methods
3.1. Molecular Modeling Study
3.2. Chemistry
3.2.1. Ethyl-2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylate: 1
3.2.2. Ethyl-2-(3-phenylthioureido)-5,6,7,8-tetrahydrocyclohexa[b]thiophene-3-carboxylate: 2
3.2.3. 3-Amino-2-phenylamino-5,6,7,8-tetrahydrocyclohexa[4,5]thieno[2,3-d]pyrimidin-4(3H)-one: 3
3.3. In Vitro Kinase Inhibition Assessment
3.4. Cell Culture
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kim, C.; Kim, B. Anti-cancer natural products and their bioactive compounds inducing ER stress-mediated apoptosis: A review. Nutrients 2018, 10, 1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baudino, T. Targeted cancer therapy: The next generation of cancer treatment. Curr. Drug Discov. Technol. 2015, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Gryshchenko, A.A.; Bdzhola, V.G.; Balanda, A.O.; Briukhovetska, N.V.; Kotey, I.M.; Golub, A.G.; Ruban, T.P.; Lukash, L.L.; Yarmoluk, S.M. Design, synthesis, and biological evaluation of N-phenylthieno[2,3-d]pyrimidin-4-amines as inhibitors of FGFR1. Bioorg. Med. Chem. 2015, 23, 2287–2293. [Google Scholar] [CrossRef] [PubMed]
- Elmongy, E.I. Thieno[2,3-d]pyrimidine derivatives: Synthetic approaches and their FLT3 kinase inhibition. J. Heterocycl. Chem. 2020, 57, 2067–2078. [Google Scholar] [CrossRef]
- Elmongy, E.I.; Khedr, M.A.; Taleb, N.A.; Awad, H.M.; Abbas, S.E.S. Design, synthesis, and biological evaluation of some cyclohepta[b]thiophene and substituted pentahydrocycloheptathieno[2,3-d]pyrimidine derivatives. J. Heterocycl. Chem. 2017, 54, 1084–1093. [Google Scholar] [CrossRef]
- Kumar, K.S.; Chamakuri, S.; Vishweshwar, P.; Iqbal, J.; Pal, M. AlCl3-induced (hetero)arylation of thienopyrimidine ring: A new synthesis of 4-substituted thieno[2,3-d]pyrimidines. Tetrahedron Lett. 2010, 51, 3269–3273. [Google Scholar] [CrossRef]
- Gorja, D.R.; Kumar, K.S.; Mukkanti, K.; Pal, M. C–C (alkynylation) vs C–O (ether) bond formation under pd/C–Cu catalysis: Synthesis and pharmacological evaluation of 4-alkynylthieno[2,3-d]pyrimidines. Beilstein J. Org. Chem. 2011, 7, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Gad, E.M.; Nafie, M.S.; Eltamany, E.H.; Hammad, M.S.A.G.; Barakat, A.; Boraei, A.T.A. Discovery of New Apoptosis-Inducing Agents for Breast Cancer Based on Ethyl 2-Amino-4,5,6,7-Tetra Hydrobenzo[b]Thiophene-3-Carboxylate: Synthesis, In Vitro, and In Vivo Activity Evaluation. Molecules 2020, 25, 2523. [Google Scholar] [CrossRef]
- Abuelhassan, S.; Bakhite, E.A.G.; Abdel-Rahman, A.E.; El-Mahdy, A.F. Synthesis, characterization, and biological activities of some novel thienylpyrido[3′,2′:4,5]thieno[3,2-d]pyrimidines and related heterocycles. J. Heterocycl. Chem. 2021, 58, 1784–1801. [Google Scholar] [CrossRef]
- Elmongy, E.I.; Attallah, N.G.M.; Altwaijry, N.; AlKahtani, M.M.; Henidi, H.A. Design and synthesis of new thiophene/thieno[2,3-d]pyrimidines along with their cytotoxic biological evaluation as tyrosine kinase inhibitors in addition to their apoptotic and autophagic induction. Molecules 2021, 27, 123. [Google Scholar] [CrossRef] [PubMed]
- El-Mekabaty, A.; Fouda, A.E.-A.S.; Shaaban, I.E.I. Convenient synthesis of functionalized thienopyrimidine-4-ones and thienopyridine-4-ones bearing a pyridine moiety with anticipated antioxidant activity. J. Heterocycl. Chem. 2020, 57, 2928–2935. [Google Scholar] [CrossRef]
- Nagaraju, K.; Bhaskaruni, V.H.; Kishore, R.; Maddila, S.; Singh, P.; Jonnalagadda, S.B. Synthesis and antioxidant evaluation of a new class of thienopyrimidinerhodanine hybrids. Lett. Drug Des. Discov. 2018, 15, 118–126. [Google Scholar] [CrossRef]
- Elmongy, E.I.; Khedr, M.A.; Taleb, N.A.; Abbas, S.E. Design and synthesis of new thienopyrimidine derivatives along with their antioxidant activity. Egypt. J. Chem. 2021, 64, 6857–6867. [Google Scholar] [CrossRef]
- Sharaky, M.; Kamel, M.; Aziz, M.A.; Omran, M.; Rageh, M.M.; Abouzid, K.A.; Shouman, S.A. Design, synthesis and biological evaluation of a new thieno[2,3-d]pyrimidine-based urea derivative with potential antitumor activity against tamoxifen sensitive and resistant breast cancer cell lines. J. Enzym. Inhib. Med. Chem. 2020, 35, 1641–1656. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Coumar, M.S.; Chu, C.Y.; Lin, W.H.; Chen, Y.R.; Chen, C.T.; Shiao, H.Y.; Rafi, S.; Wang, S.Y.; Hsu, H.; et al. Design and synthesis of tetrahydropyridothieno[2,3-d]pyrimidine scaffold based epidermal growth factor receptor (EGFR) kinase inhibitors: The role of side chain chirality and michael acceptor group for maximal potency. J. Med. Chem. 2010, 53, 7316–7326. [Google Scholar] [CrossRef]
- Heng, H.; Zhi, Y.; Yuan, H.; Wang, Z.; Li, H.; Wang, S.; Tian, J.; Liu, H.; Chen, Y.; Lu, T.; et al. Discovery of a highly selective FLT3 inhibitor with specific proliferation inhibition against AML cells harboring FLT3-ITD mutation. Eur. J. Med. Chem. 2019, 163, 195–206. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Holland, S.M.; Staudt, L.M. Mechanisms of disease: JAKs and STATs in immunity, immunodeficiency, and cancer. N. Engl. J. Med. 2013, 368, 161–170. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [Green Version]
- Hart, S.; Goh, K.C.; Novotny-Diermayr, V.; Hu, C.Y.; Hentze, H.; Tan, Y.C.; Madan, B.; Amalini, C.; Loh, Y.K.; Ong, L.C.; et al. SB1518, a novel macrocyclic pyrimidine-based JAK2 inhibitor for the treatment of myeloid and lymphoid malignancies. Leukemia 2011, 25, 1751–1759. [Google Scholar] [CrossRef] [Green Version]
- Pesu, M.; Laurence, A.; Kishore, N.; Zwillich, S.H.; Chan, G.; O’Shea, J.J. Therapeutic targeting of janus kinases. Immunol. Rev. 2008, 223, 132–142. [Google Scholar] [CrossRef] [Green Version]
- Norman, P. Selective JAK inhibitors in development for rheumatoid arthritis. Expert Opin. Investig. Drugs 2014, 23, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Kontzias, A.; Kotlyar, A.; Laurence, A.; Changelian, P.; O’Shea, J.J. Jakinibs: A new class of kinase inhibitors in cancer and autoimmune disease. Curr. Opin. Pharmacol. 2012, 12, 464–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talpaz, M.; Kiladjian, J. Fedratinib, a newly approved treatment for patients with myeloproliferative neoplasm-associated myelofibrosis. Leukemia 2021, 35, 1–17. [Google Scholar] [CrossRef]
- Mesa, R.A. Ruxolitinib, a selective JAK1 and JAK2 inhibitor for the treatment of myeloproliferative neoplasms and psoriasis. IDrugs Investig. Drugs J. 2010, 13, 394–403. [Google Scholar]
- Cingolani, A.; Tummolo, A.M.; Montemurro, G.; Gremese, E.; Larosa, L.; Cipriani, M.C.; Pasciuto, G.; Liperoti, R.; Murri, R.; Pirronti, T.; et al. Baricitinib as rescue therapy in a patient with COVID-19 with no complete response to sarilumab. Infection 2020, 48, 767–771. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Kontzias, A.; Yamaoka, K.; Tanaka, Y.; Laurence, A. Janus kinase inhibitors in autoimmune diseases. Ann. Rheum. Dis. 2013, 72, ii111–ii115. [Google Scholar] [CrossRef]
- Gupta, M.; Lee, H.J.; Barden, C.J.; Weaver, D.F. The Blood–Brain barrier (BBB) score. J. Med. Chem. 2019, 62, 9824–9836. [Google Scholar] [CrossRef]
- Khan, O.A.; Gore, M.; Lorigan, P.; Stone, J.; Greystoke, A.; Burke, W.; Carmichael, J.; Watson, A.J.; McGown, G.; Thorncroft, M.; et al. A phase I study of the safety and tolerability of olaparib (AZD2281, KU0059436) and dacarbazine in patients with advanced solid tumours. Br. J. Cancer 2011, 104, 750–755. [Google Scholar] [CrossRef] [Green Version]
- Elmongy, E.I.; Altwaijry, N.; Attallah, N.G.M.; AlKahtani, M.M.; Henidi, H.A. In-Silico Screening of Novel Synthesized Thienopyrimidines Targeting Fms Related Receptor Tyrosine Kinase-3 and Their In-Vitro Biological Evaluation. Pharmaceuticals 2022, 15, 170. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein PDB Code | E | RMSD (A) | Co-Crystalized Ligand Amino Acid Interactions | Amino Acids Involved in Interactions by the Prepared Compound |
|---|---|---|---|---|
| 5AEP | −6.3092 | 1.1599 | Arg 980, Glu 930, Leu 932, Leu 855 | Arg 980, Glu 930, Leu 855 |
| 4C62 | −5.239 | 1.9175 | Leu 932, Leu 855 | Leu 932 |
| 3ZMM | −6.0183 | 1.7528 | Glu 930, Leu 932 | Leu 932, Glu 930, Leu 855, Gly 935 |
| HT-29 | HepG-2 | MCF-7 | |
|---|---|---|---|
| Compound 3 | 4.526 ± 0.130 | 8.001 ± 0.0445 | 15.055 ± 0.785 |
| Doxorubicin | 1.358 ± 1.156 | 13.91 ± 2.170 | 8.434 ± 0.522 |
| Compound | Molecular Weight | Number of HBA | Number of HBD | Log P | MR | TPSA | BBB Score | GI Absorption | Drug Likeness Score | Compounds in Relation to Drug and Non-Drug Scores |
|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 312.39 | 2 | 2 | 2.63 | 91 | 101.18 | 4.01 | High | 0.57 |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elmongy, E.I.; Henidi, H.A. In Silico Evaluation of a Promising Key Intermediate Thieno [2,3-d] Pyrimidine Derivative with Expected JAK2 Kinase Inhibitory Activity. Molbank 2022, 2022, M1352. https://doi.org/10.3390/M1352
Elmongy EI, Henidi HA. In Silico Evaluation of a Promising Key Intermediate Thieno [2,3-d] Pyrimidine Derivative with Expected JAK2 Kinase Inhibitory Activity. Molbank. 2022; 2022(1):M1352. https://doi.org/10.3390/M1352
Chicago/Turabian StyleElmongy, Elshaymaa I., and Hanan Ali Henidi. 2022. "In Silico Evaluation of a Promising Key Intermediate Thieno [2,3-d] Pyrimidine Derivative with Expected JAK2 Kinase Inhibitory Activity" Molbank 2022, no. 1: M1352. https://doi.org/10.3390/M1352
APA StyleElmongy, E. I., & Henidi, H. A. (2022). In Silico Evaluation of a Promising Key Intermediate Thieno [2,3-d] Pyrimidine Derivative with Expected JAK2 Kinase Inhibitory Activity. Molbank, 2022(1), M1352. https://doi.org/10.3390/M1352