
4,4’-(Pyridin-4-ylmethylene)dibenzonitrile



Abstract
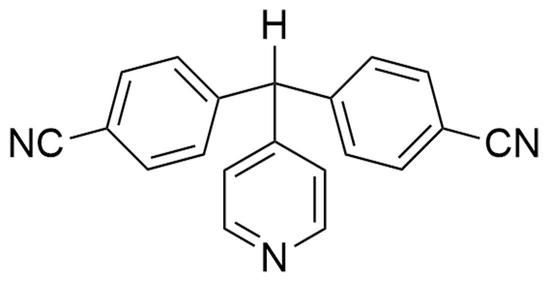
1. Introduction
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bunnett, J.F.; Zahler, R.E. Aromatic Nucleophilic Substitution Reactions. Chem. Rev. 1951, 49, 273–412. [Google Scholar] [CrossRef]
- Rohrbach, S.; Smith, A.J.; Pang, J.H.; Poole, D.L.; Tuttle, T.; Chiba, S.; Murphy, J.A. Concerted Nucleophilic Aromatic Substitution Reactions. Angew. Chem. 2019, 58, 16368–16388. [Google Scholar] [CrossRef] [PubMed]
- Bella, M.; Kobbelgaard, S.; Jørgensen, K.A. Organocatalytic regio- and asymmetric C-selective S(N)Ar reactions-stereoselective synthesis of optically active spiro-pyrrolidone-3,3′-oxoindoles. J. Am. Chem. Soc. 2005, 127, 3670–3671. [Google Scholar] [CrossRef] [PubMed]
- Arita, M.; Yokoyama, S.; Asahara, H.; Nishiwaki, N. Three Step Synthesis of Fully and Differently Arylated Pyridines. Eur. J. Org. Chem. 2020, 31, 466–474. [Google Scholar] [CrossRef]
- Stefanidis, D.; Bunting, J.W. Rate-equilibrium relationships for the deprotonation of 4-phenacylpyridines and 4-phenacylpyridinium cations. J. Am. Chem. 1990, 112, 3163–3168. [Google Scholar] [CrossRef]
- Chen, H.Y.; Kim, S.; Wu, J.Y.; Birzin, E.T.; Chan, W.; Yang, Y.T.; Dahllund, J.; DiNinno, F.; Rohrer, S.P.; Schaeffer, J.M.; et al. Estrogen receptor ligands. Part 3: The SAR of dihydrobenzoxathiin SERMs. Bioorg. Med. Chem. Lett. 2004, 14, 2551–2554. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Baxendale, I.R. Continuous-Flow Synthesis of 2H-Azirines and Their Diastereoselective Transformation to Aziridines. Synlett 2016, 27, 159–163. [Google Scholar] [CrossRef][Green Version]
- Utsugi, Y.; Kobuchi, H.; Kawamura, Y.; Atito, A.S.A.; Nagao, M.; Isoda, H.; Miyamae, Y. Importance of the proximity and orientation of ligand-linkage to the design of cinnamate-GW9662 hybrid compounds as covalent PPAR agonists. Molecules 2019, 24, 2019. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.; Chae, B.; Seo, H.; Jung, Y.M.; Lee, S.W. Structural characterization of triphenylamine (TPA)-based polymers during the oxidative reaction by two-dimensional (2D) infrared correlation study. J. Mol. Struct. 2014, 1069, 200–204. [Google Scholar] [CrossRef]
- Wilshire, J.J.F.K.J.; Trantino, G.G.J.G.; Mackay, M.; Trantino, G.G.J.G.; Wilshire, J.J.F.K.J. The Reaction of Some N-(Nitrophenyl)azoles With Alkali: Preparation of the Corresponding Azoxybenzenes. X-Ray Structure of 2, 2′-Bis(1″,2″,4″-triazol-1″-yl)azoxybenzene. Aust. J. Chem. 1993, 46, 417–425. [Google Scholar]
- Nicholls, A.J.; Baxendale, I.R. Benzo[1,2,3]dithiazole Compounds: A History of Synthesis and Their Renewed Applicability in Materials and Synthetic Chemistry, Originating from the Herz Reaction. Reactions 2021, 2, 175–208. [Google Scholar] [CrossRef]
- Mente, S.; Arnold, E.; Butler, T.; Chakrapani, S.; Chandrasekaran, R.; Cherry, K.; Dirico, K.; Doran, A.; Fisher, K.; Galatsis, P.; et al. Ligand-protein interactions of selective casein kinase 1δ inhibitors. J. Med. Chem. 2013, 56, 6819–6828. [Google Scholar] [CrossRef] [PubMed]
- Gadsby, J.M.; McPake, C.B.; Murray, C.B.; Sandford, G. Synthesis of polyfluorinated terphenyl and styrene derivatives by palladium catalysed C-F bond activation of polyfluoronitroaromatic substrates. J. Fluor. Chem. 2016, 181, 51–55. [Google Scholar] [CrossRef][Green Version]
- Zhang, S.; Kim, B.S.; Wu, C.; Mao, J.; Walsh, P.J. Palladium-catalysed synthesis of triaryl(heteroaryl)methanes. Nat. Commun. 2017, 8, 14641. [Google Scholar] [CrossRef] [PubMed]
- Seerden, J.-P.G.; Leusink-Ionescu, G.; Leguijt, R.; Saccavini, C.; Gelensa, E.; Dros, B.; Woudenberg-Vrenken, T.; Molema, G.; Kamps, J.A.A.M.; Kellogg, R.M. Syntheses and structure–activity relationships for some triazolyl p38α MAPK inhibitors. Bioorganic Med. Chem. Lett. 2014, 24, 1352–1357. [Google Scholar] [CrossRef] [PubMed]
- Nabavizadeh, S.M.; Rashidi, M. Acidity of osmium tetroxide (OsO4) towards coordination with pyridine and its derivatives. Polyhedron 2007, 26, 1476–1482. [Google Scholar] [CrossRef]
- Calculated using Advanced Chemistry Development (ACD/Labs) Software V11.02. Available online: https://www.acdlabs.com/products/percepta/predictors/pka/index.php (accessed on 19 November 2021).
- Sterckx, H.; Houwer, J.D.; Mensch, C.; Herrebout, W.; Tehrani, K.A.; Maes, B.U.W. Base metal-catalyzed benzylic oxidation of (aryl)(heteroaryl)methanes with molecular oxygen. Beilstein J. Org. Chem. 2016, 12, 144–153. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lancaster, B.M.J.; Nicholls, A.J.; Baxendale, I.R. 4,4’-(Pyridin-4-ylmethylene)dibenzonitrile. Molbank 2021, 2021, M1302. https://doi.org/10.3390/M1302
Lancaster BMJ, Nicholls AJ, Baxendale IR. 4,4’-(Pyridin-4-ylmethylene)dibenzonitrile. Molbank. 2021; 2021(4):M1302. https://doi.org/10.3390/M1302
Chicago/Turabian StyleLancaster, Ben M. J., Alexander J. Nicholls, and Ian R. Baxendale. 2021. "4,4’-(Pyridin-4-ylmethylene)dibenzonitrile" Molbank 2021, no. 4: M1302. https://doi.org/10.3390/M1302
APA StyleLancaster, B. M. J., Nicholls, A. J., & Baxendale, I. R. (2021). 4,4’-(Pyridin-4-ylmethylene)dibenzonitrile. Molbank, 2021(4), M1302. https://doi.org/10.3390/M1302