
2-Oxo-2H-chromen-7-yl 4-chlorobenzoate


Abstract
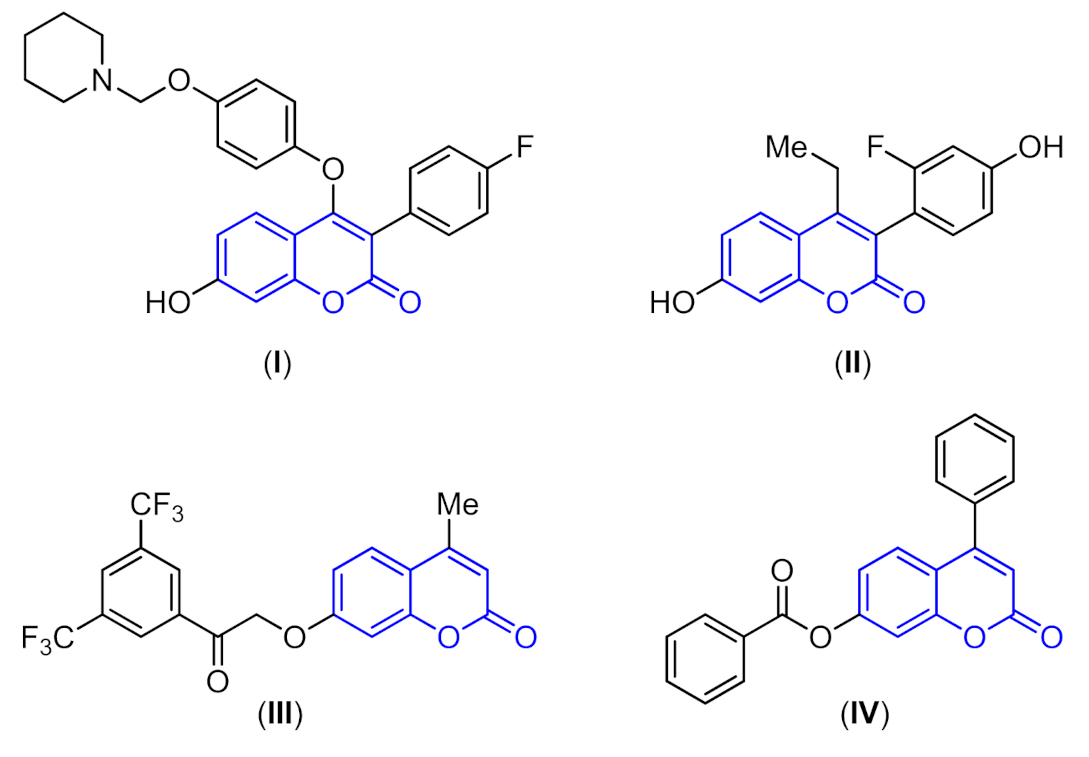
:1. Introduction
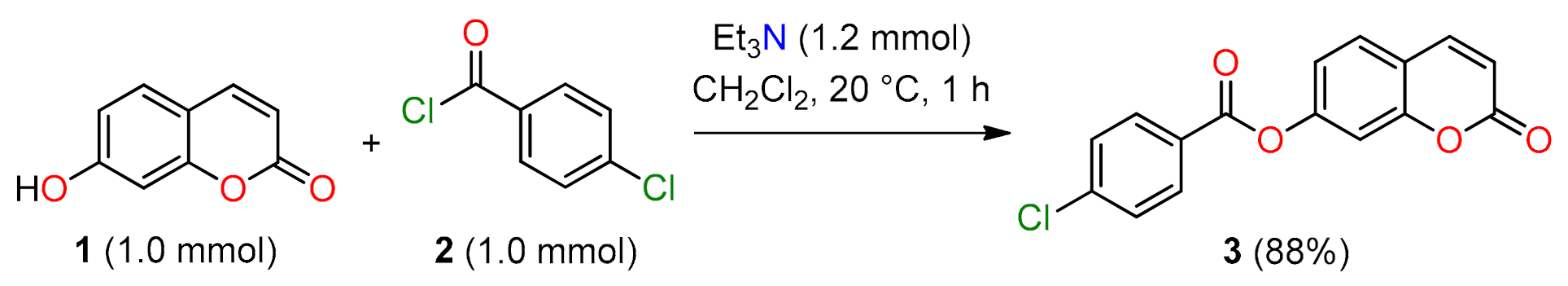
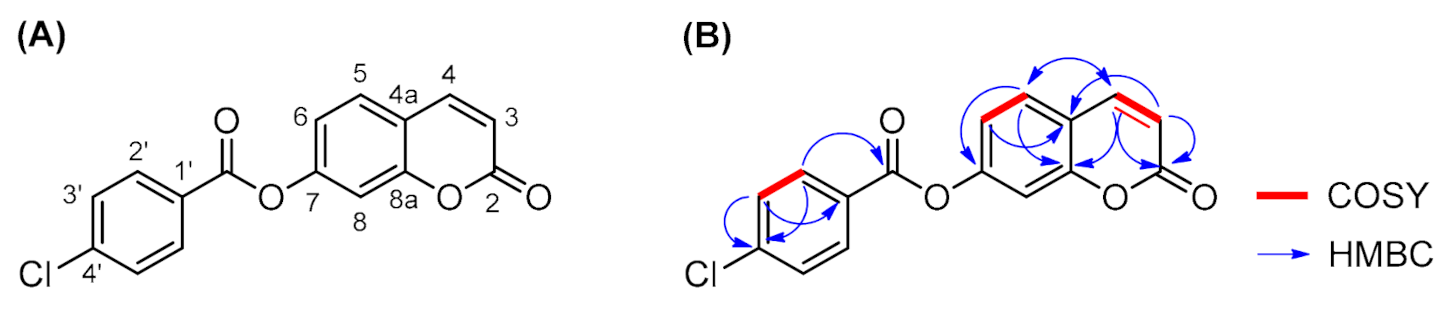
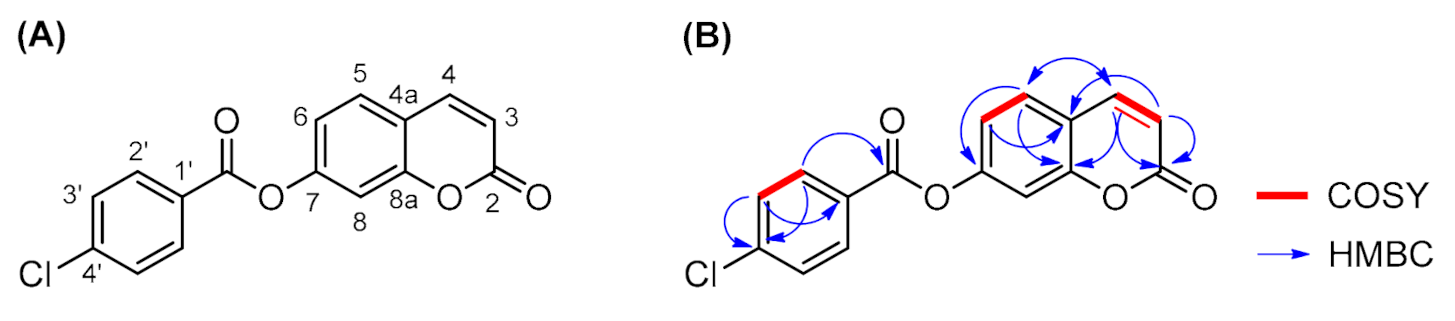
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthesis of 2-Oxo-2H-Chromen-7-yl 4-Chlorobenzoate 3
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vogel, A. Darftellung von benzoefäure aus der tonka-bohns und aus den meliloten-oder steinklee-blumen. Ann. Phys. 1820, 64, 161–166. [Google Scholar] [CrossRef] [Green Version]
- Perkin, W.H. VI.—On the artificial production of coumarin and formation of its homologues. J. Chem. Soc. 1868, 21, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Cao, D.; Liu, Z.; Verwilst, P.; Koo, S.; Jangjili, P.; Kim, J.S.; Lin, W. Coumarin-based small-molecule fluorescent chemosensors. Chem. Rev. 2019, 119, 10403–10519. [Google Scholar] [CrossRef] [PubMed]
- Grover, J.; Jachak, S.M. Coumarins as privileged scaffold for anti-inflammatory drug development. RSC Adv. 2015, 5, 38892–38905. [Google Scholar] [CrossRef]
- Insuasty, D.; Castillo, J.; Becerra, D.; Rojas, H.; Abonia, R. Synthesis of biologically active molecules through multicomponent reactions. Molecules 2020, 25, 505. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Tan, X.; Liang, C.; Zhang, W. Design, synthesis, and antifungal evaluation of novel coumarin-pyrrole hybrids. J. Heterocycl. Chem. 2021, 58, 450–458. [Google Scholar] [CrossRef]
- Venugopala, K.N.; Rashmi, V.; Odhav, B. Review on natural coumarin lead compounds for their pharmacological activity. Biomed. Res. Int. 2013, 2013, 963248. [Google Scholar] [CrossRef] [Green Version]
- Katsori, A.-M.; Hadjipavlou-Litina, D. Coumarin derivatives: An updated patent review (2012–2014). Expert Opin. Ther. Patents 2014, 24, 1323–1347. [Google Scholar] [CrossRef]
- Hassan, M.Z.; Osman, H.; Ali, M.A.; Ahsan, M.J. Therapeutic potential of coumarins as antiviral agents. Eur. J. Med. Chem. 2016, 123, 236–255. [Google Scholar] [CrossRef]
- Stefanachi, A.; Leonetti, F.; Pisani, L.; Catto, M.; Carotti, A. Coumarin: A natural, privileged and versatile scaffold for bioactive compounds. Molecules 2018, 23, 250. [Google Scholar] [CrossRef] [Green Version]
- Musa, A.M.; Cooperwood, J.S.; Khan, M.O.F. A review of coumarin derivatives in pharmacotherapy of breast cancer. Curr. Med. Chem. 2008, 15, 2664–2679. [Google Scholar] [CrossRef] [Green Version]
- Thakur, A.; Singla, R.; Jaitak, V. Coumarins as anticancer agents: A review on synthetic strategies, mechanism of action and SAR studies. Eur. J. Med. Chem. 2015, 101, 476–495. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, Z. Coumarin-containing hybrids and their anticancer activities. Eur. J. Med. Chem. 2019, 181, 111587. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, J.; Liu, Y.; Zeng, Y.; Wu, G. A review on anti-tumor mechanisms of coumarins. Front. Oncol. 2020, 10, 592853. [Google Scholar] [CrossRef]
- Al-Warhi, T.; Sabt, A.; Elkaeed, E.B.; Eldehna, W.M. Recent advancements of coumarin-based anticancer agents: An up-to-date review. Bioorg. Chem. 2020, 103, 104163. [Google Scholar] [CrossRef]
- Kandil, S.; Westwell, A.D.; McGuigan, C. 7-Substituted umbelliferone derivatives as androgen receptor antagonists for the potential treatment of prostate and breast cancer. Bioorg. Med. Chem. Lett. 2016, 26, 2000–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meleddu, R.; Deplano, S.; Maccioni, E.; Ortuso, F.; Cottiglia, F.; Secci, D.; Onali, A.; Sanna, E.; Angeli, A.; Angius, R.; et al. Selective inhibition of carbonic anhydrase IX and XII by coumarin and psoralen derivatives. J. Enzyme Inhib. Med. Chem. 2021, 36, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Medina, F.G.; Gonzalez-Marrero, J.; Macías-Alonso, M.; González, M.C.; Córdova-Guerrero, I.; García, A.G.T.; Osegueda-Robles, S. Coumarin heterocyclic derivatives: Chemical synthesis and biological activity. Nat. Prod. Rep. 2015, 32, 1472–1507. [Google Scholar] [CrossRef]
- Cui, J.; Li, M.-L.; Yuan, M.-S. Antifeedant activities of tutin and 7-hydroxycoumarin acylation derivatives against Mythimna separate. J. Pestic. Sci. 2012, 37, 95–98. [Google Scholar] [CrossRef] [Green Version]
- Ji, W.; Li, L.; Eniola-Adefeso, O.; Wang, Y.; Liu, C.; Feng, C. Non-invasively visualizing cell–matrix interactions in two-photon excited supramolecular hydrogels. J. Mater. Chem. B 2017, 5, 7790–7795. [Google Scholar] [CrossRef]
- Orhan, I.E.; Deniz, S.S.; Salmas, R.E.; Durdagi, S.; Epifano, F.; Genovese, S.; Fiorito, S. Combined molecular modeling and cholinesterase inhibition studies on some natural and semisynthetic O-alkylcoumarin derivatives. Bioorg. Chem. 2019, 84, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.-C.; Bravo, N.-F.; Tamayo, L.-V.; Mestizo, P.-D.; Hurtado, J.; Macías, M.; Portilla, J. Water-compatible synthesis of 1,2,3-triazoles under ultrasonic conditions by a Cu(I) complex-mediated click reaction. ACS Omega 2020, 5, 30148–30159. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Fuquen, R.; Arango-Daraviña, K.; Becerra, D.; Castillo, J.-C.; Kennedy, A.R.; Macías, M.A. Catalyst- and solvent-free synthesis of 2-fluoro-N-(3-methyl sulfanyl-1H-1,2,4-triazol-5-yl)benzamide through a microwave-assisted Fries rearrangement: X-ray structural and theoretical studies. Acta Crystallogr. Sect. C Struct. Chem. 2019, 75, 359–371. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Fuquen, R.; Hincapié-Otero, M.M.; Becerra, D.; Castillo, J.-C.; Portilla, J.; Macías, M.A. Synthesis of 1-aroyl-3-methylsulfanyl-5-amino-1,2,4-triazoles and their analysis by spectroscopy, X-ray crystallography and theoretical calculations. J. Mol. Struct. 2021, 1226, 129317. [Google Scholar] [CrossRef]
- Shakila, G.; Periandy, S.; Ramalingam, S. Molecular structure and vibrational analysis of 1-bromo-2-chlorobenzene using ab initio HF and density functional theory (B3LYP) calculations. J. At. Mol. Opt. Phys. 2011, 2011, 512841. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Number | δH (mult, J in Hz) | δC (ppm) | COSY (1H-1H) | NOESY (1H-1H) | HMBC (1H-13C) |
|---|---|---|---|---|---|
| 2 | -- | 159.7 | -- | -- | H-3 (2J) H-4 (3J) |
| 3 | 6.51 (d, J = 9.6) | 115.8 | H-4 (3J) | H-4 | -- |
| 4 | 8.11 (d, J = 9.6) | 143.9 | H-3 (3J) | H-3 H-5 | H-5 (3J) |
| 4a | -- | 117.0 | -- | -- | H-3 (3J) H-6 (3J) |
| 5 | 7.83 (d, J = 8.4) | 129.5 | H-6 (3J) | H-4 H-6 | H-4 (3J) |
| 6 | 7.34 (dd, J = 8.4, 2.0) | 118.8 | H-5 (3J) | H-5 | -- |
| 7 | -- | 152.9 | -- | -- | H-5 (3J) |
| 8 | 7.48 (d, J = 2.0) | 110.4 | -- | -- | -- |
| 8a | -- | 154.1 | -- | -- | H-4 (3J) H-5 (3J) |
| 1′ | -- | 127.4 | -- | -- | H-3′ (3J) |
| 2′ | 8.15 (d, J = 8.4) | 131.8 | H-3′ (3J) | H-3′ | -- |
| 3′ | 7.70 (d, J = 8.4) | 129.2 | H-2′ (3J) | H-2′ | -- |
| 4′ | -- | 139.3 | -- | -- | H-2′ (3J) H-3′ (2J) |
| C=O | -- | 163.4 | -- | -- | H-2′ (3J) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becerra, D.; Portilla, J.; Castillo, J.-C. 2-Oxo-2H-chromen-7-yl 4-chlorobenzoate. Molbank 2021, 2021, M1279. https://doi.org/10.3390/M1279
Becerra D, Portilla J, Castillo J-C. 2-Oxo-2H-chromen-7-yl 4-chlorobenzoate. Molbank. 2021; 2021(3):M1279. https://doi.org/10.3390/M1279
Chicago/Turabian StyleBecerra, Diana, Jaime Portilla, and Juan-Carlos Castillo. 2021. "2-Oxo-2H-chromen-7-yl 4-chlorobenzoate" Molbank 2021, no. 3: M1279. https://doi.org/10.3390/M1279
APA StyleBecerra, D., Portilla, J., & Castillo, J.-C. (2021). 2-Oxo-2H-chromen-7-yl 4-chlorobenzoate. Molbank, 2021(3), M1279. https://doi.org/10.3390/M1279