
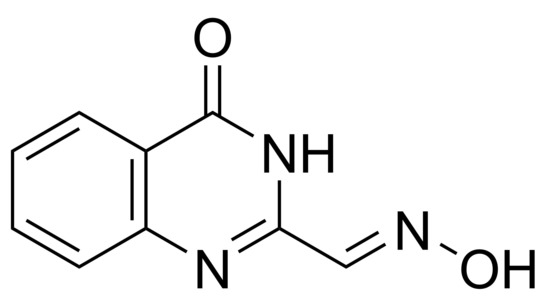
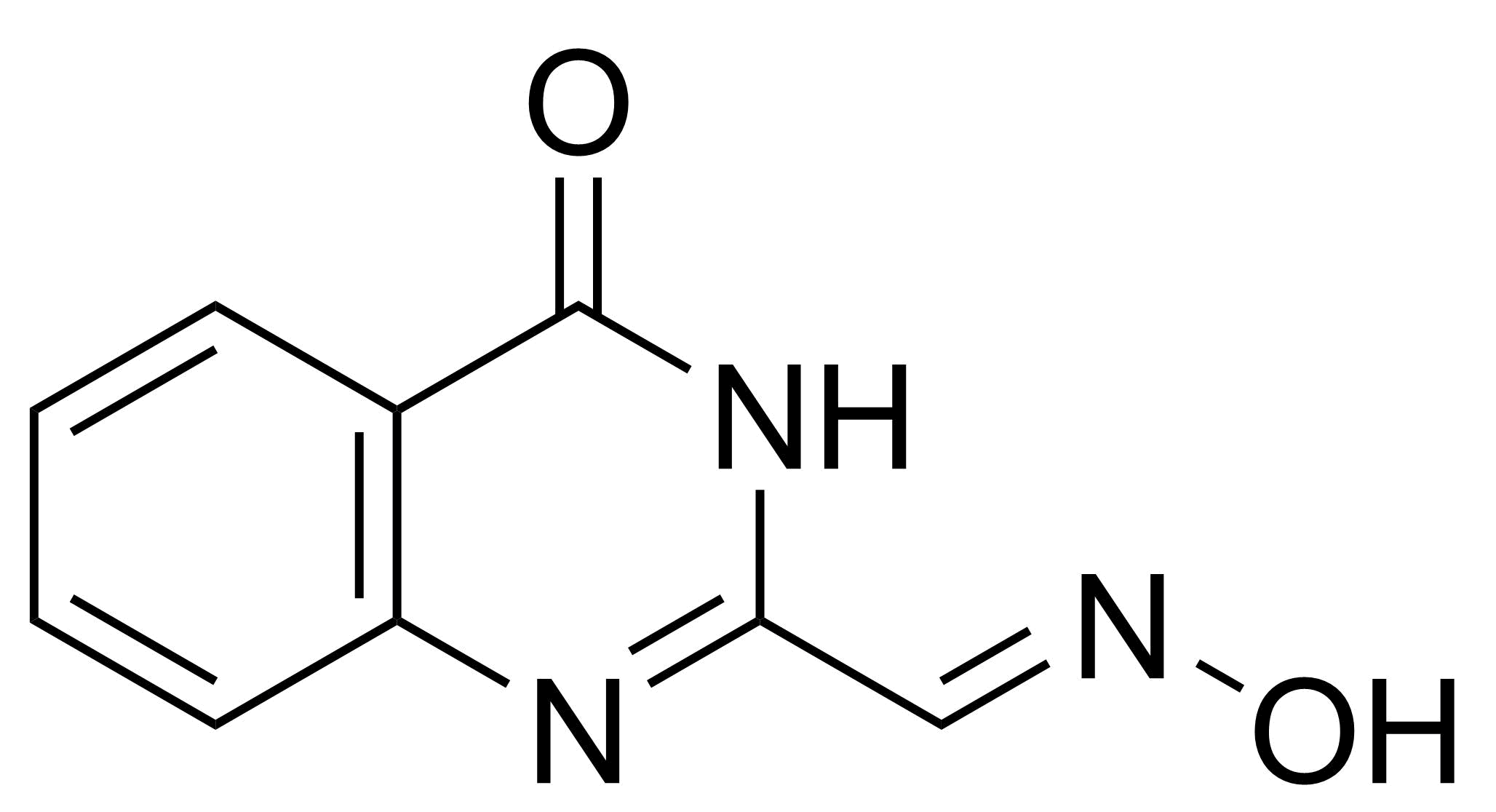
(E)-4-Oxo-3,4-dihydroquinazoline-2-carbaldehyde Oxime



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
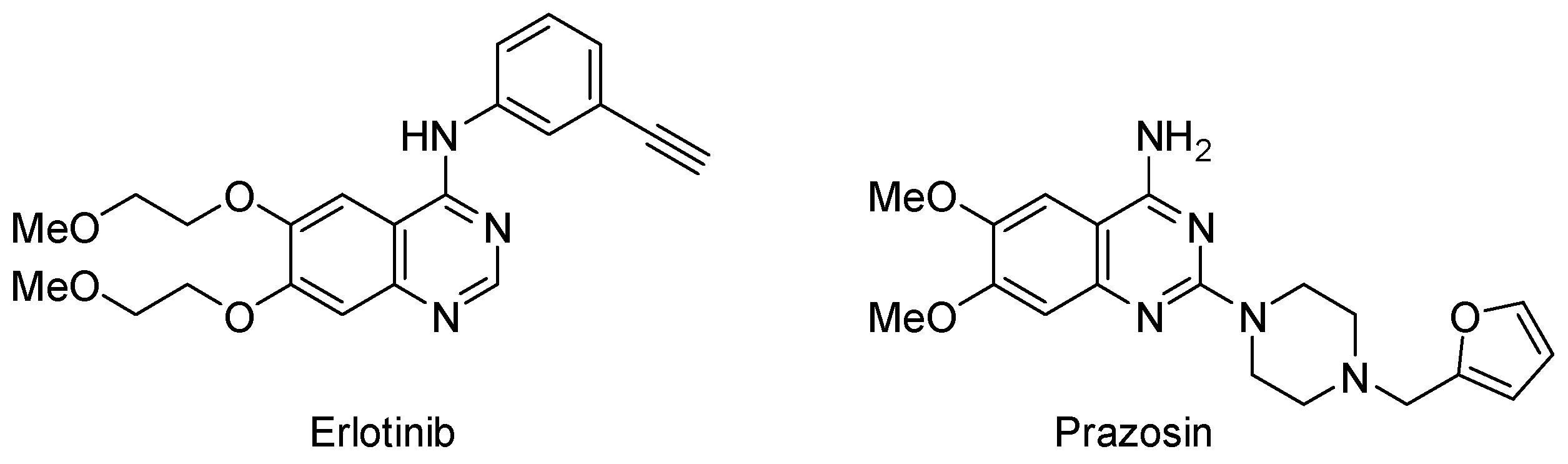
1. Introduction
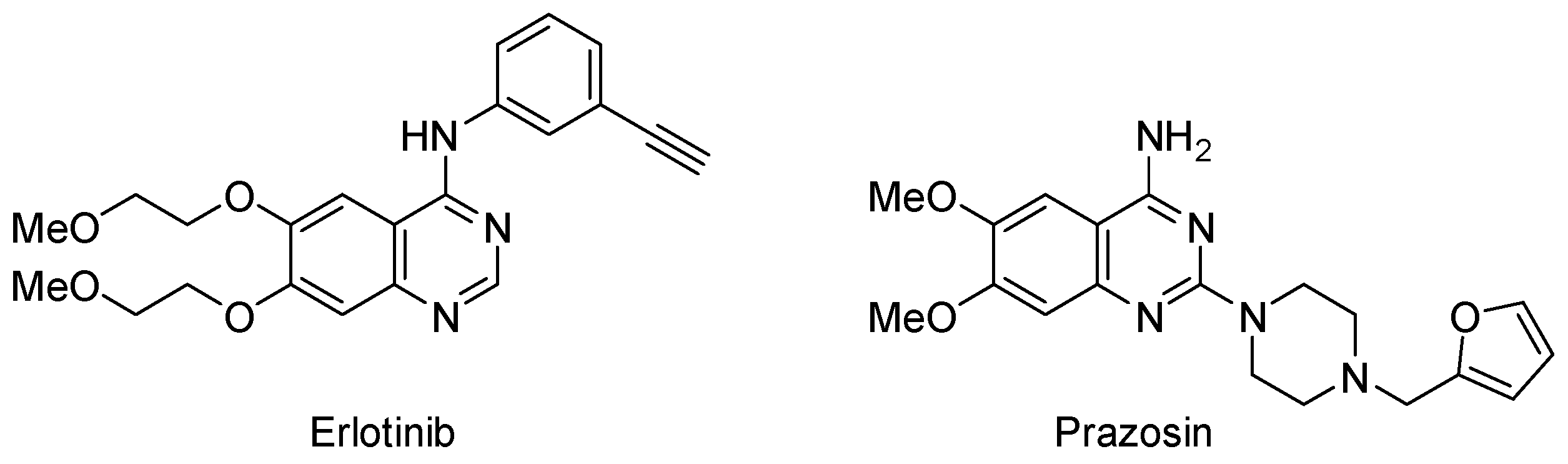
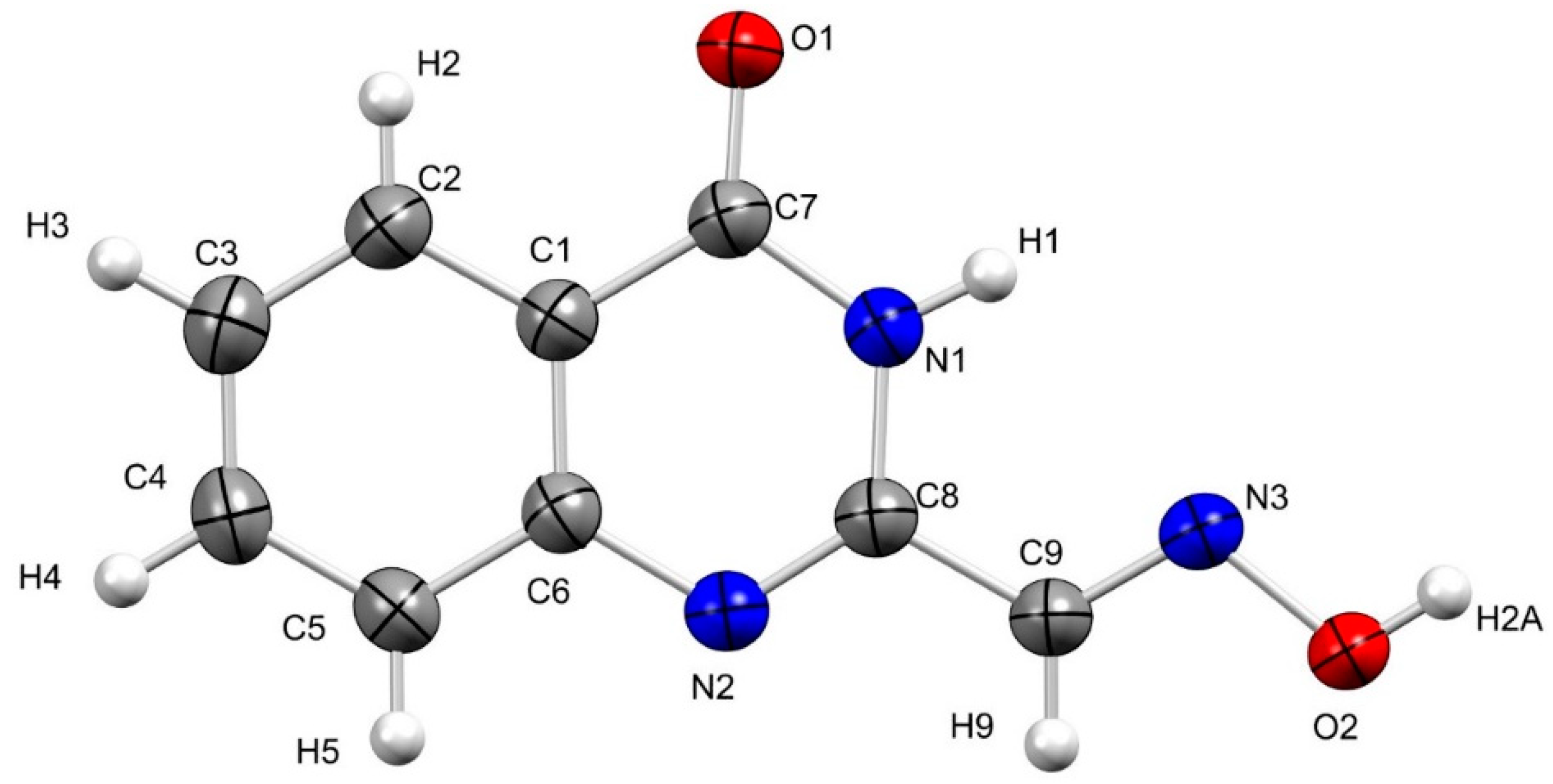
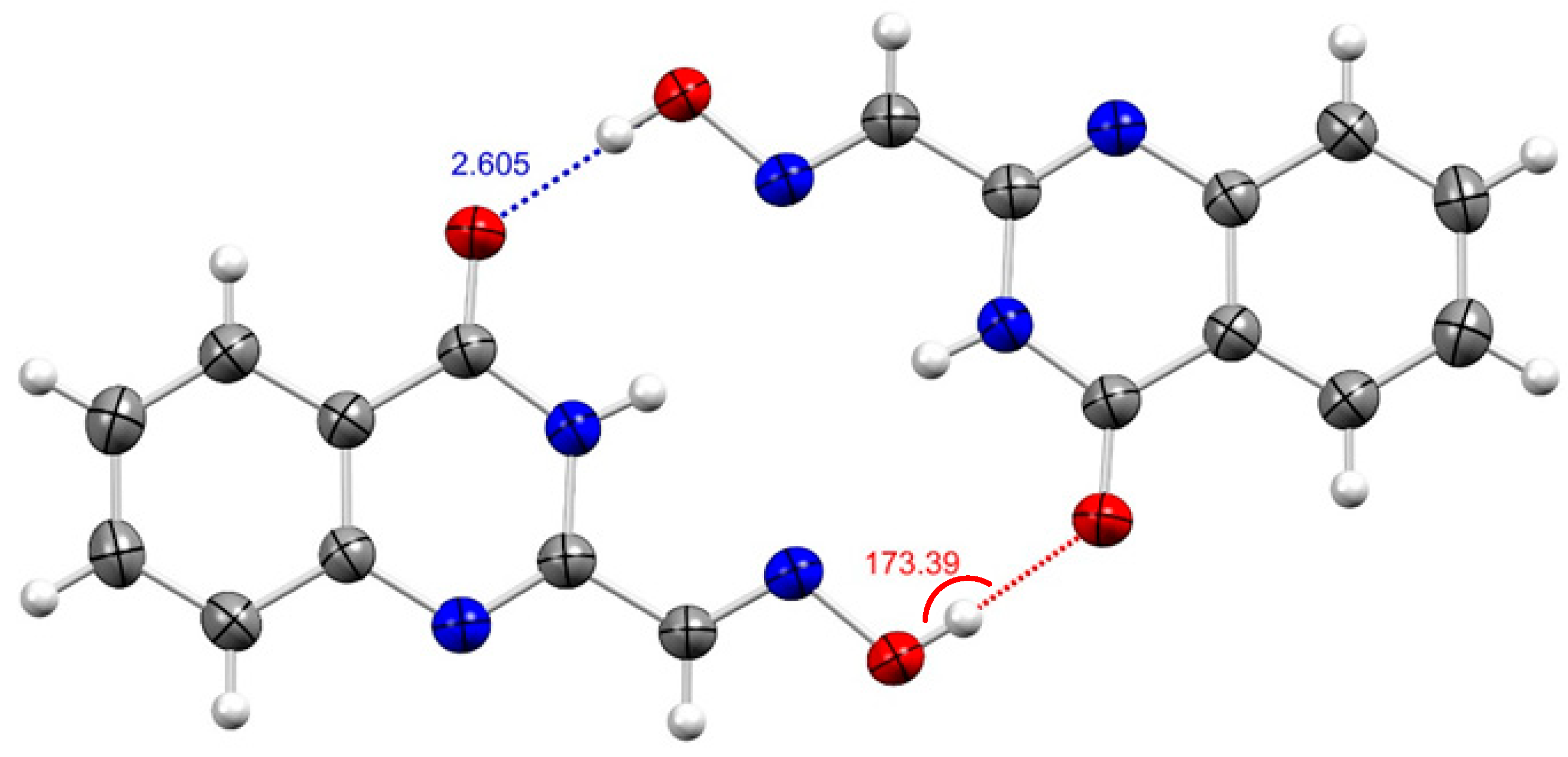
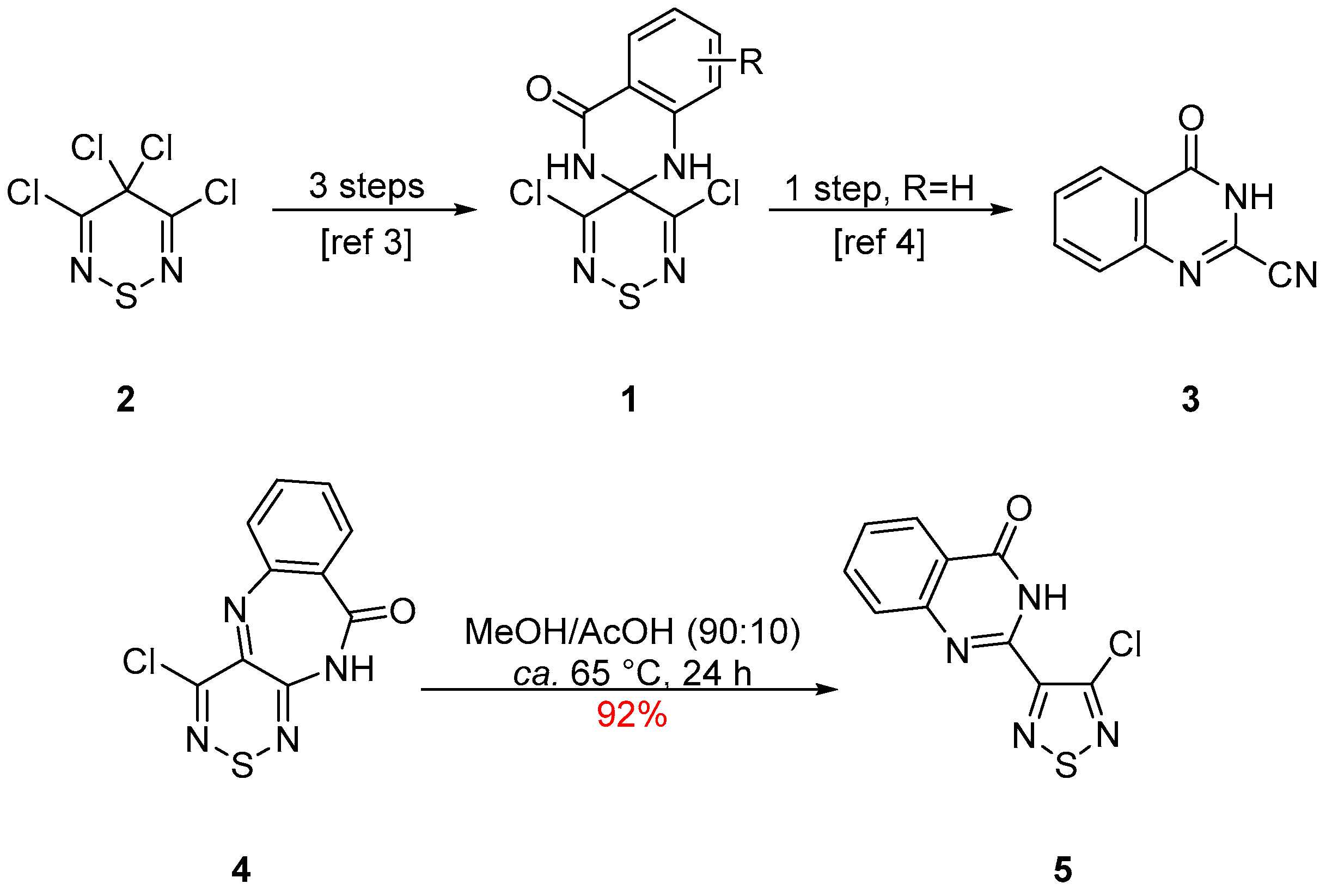
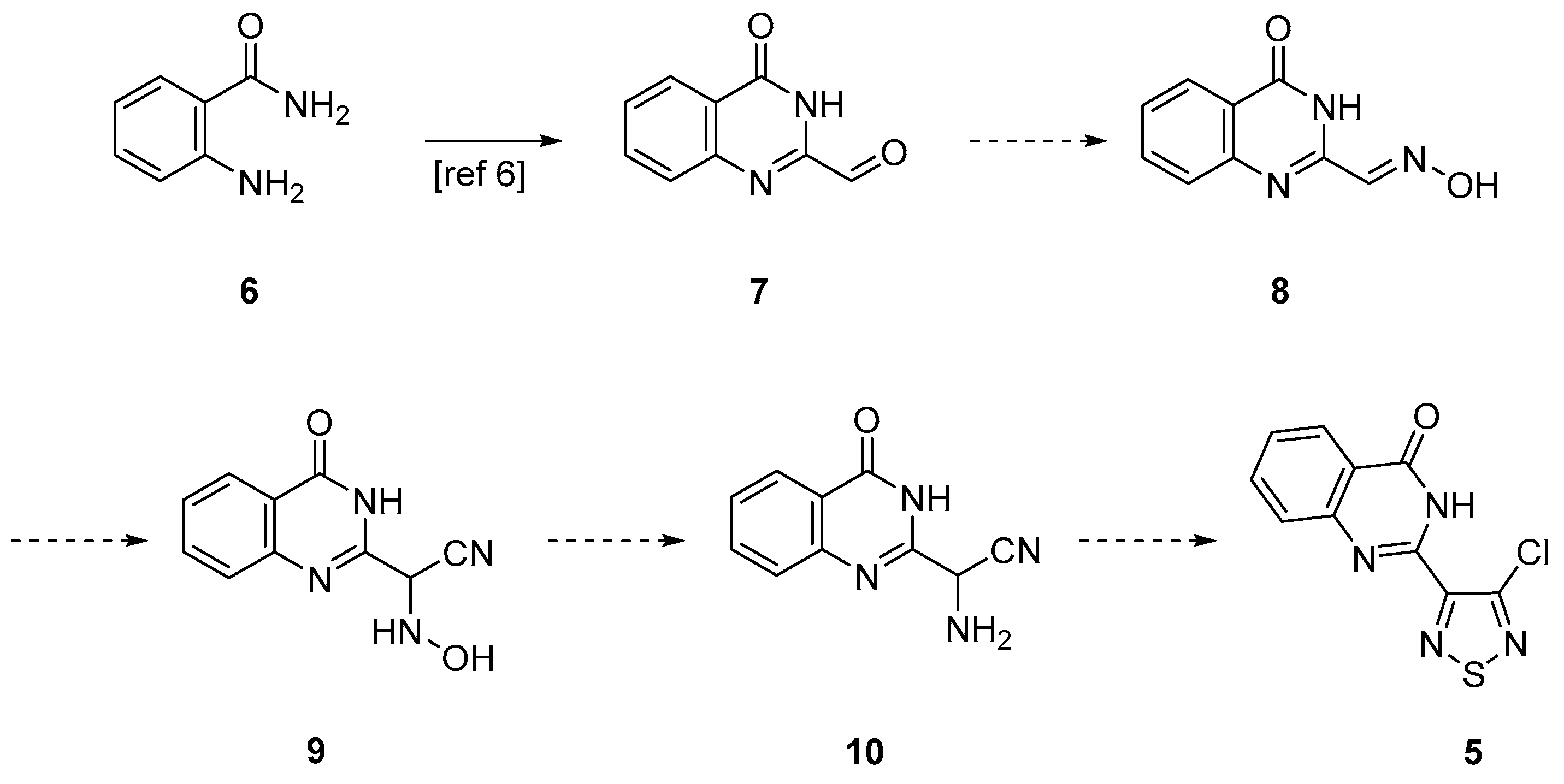
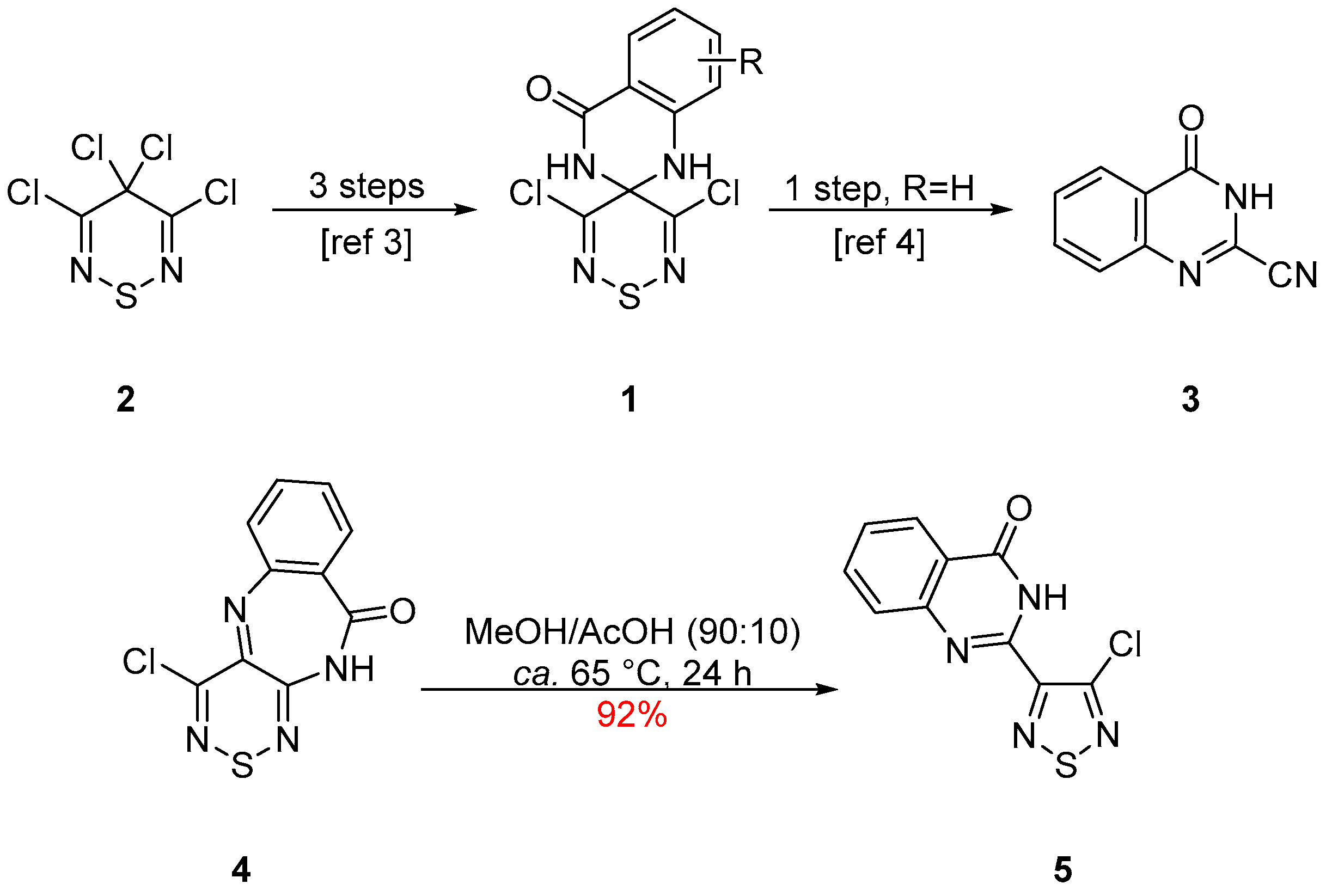
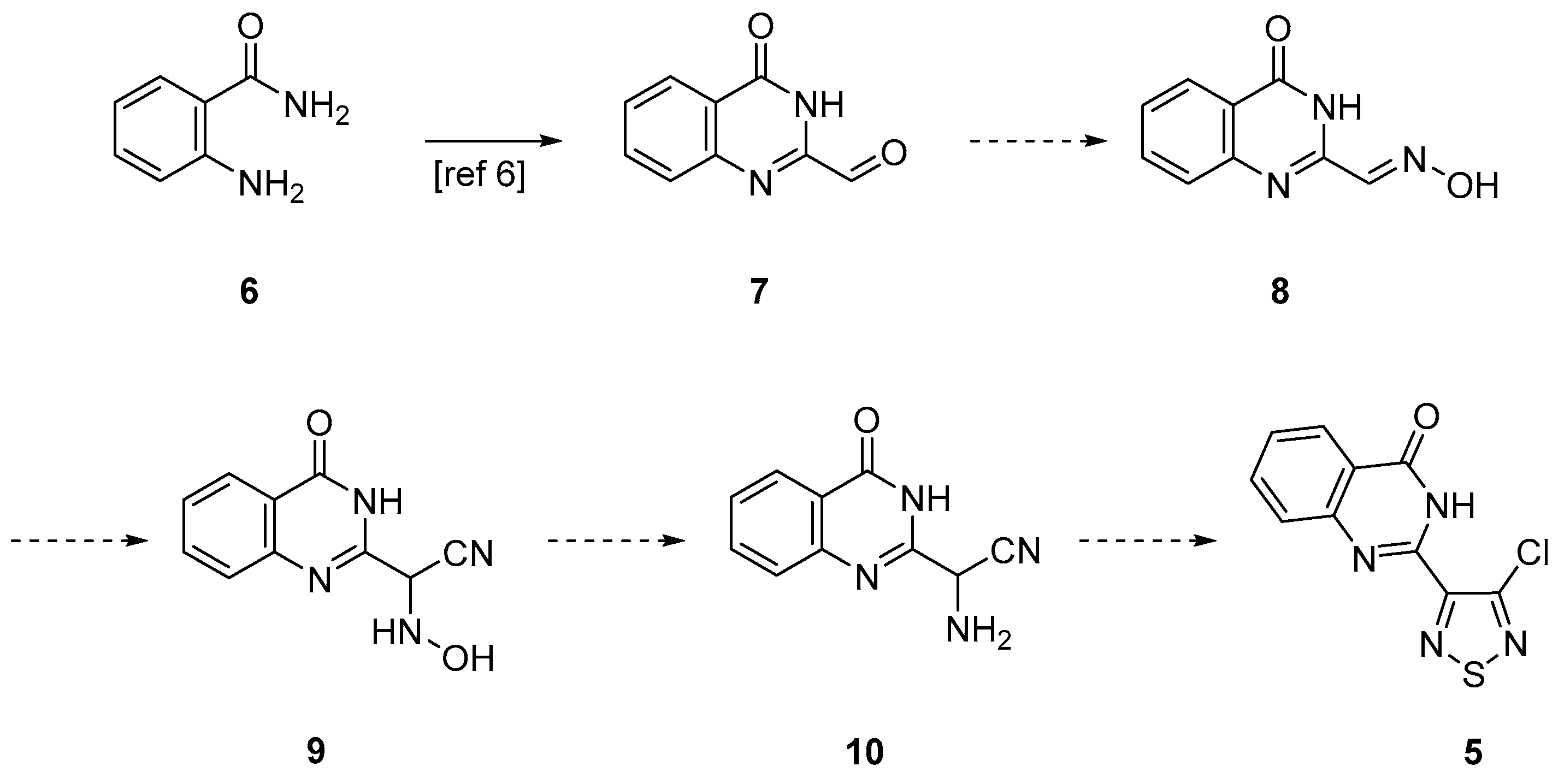
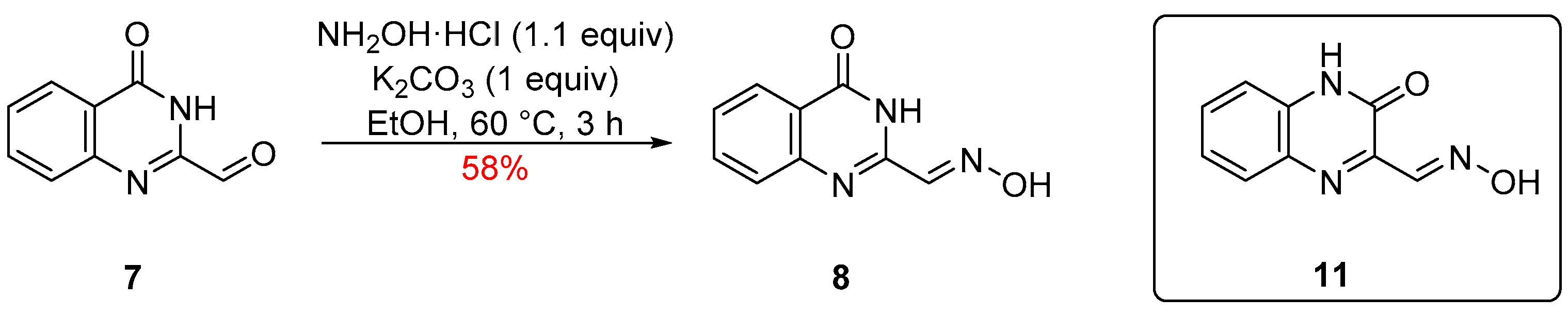
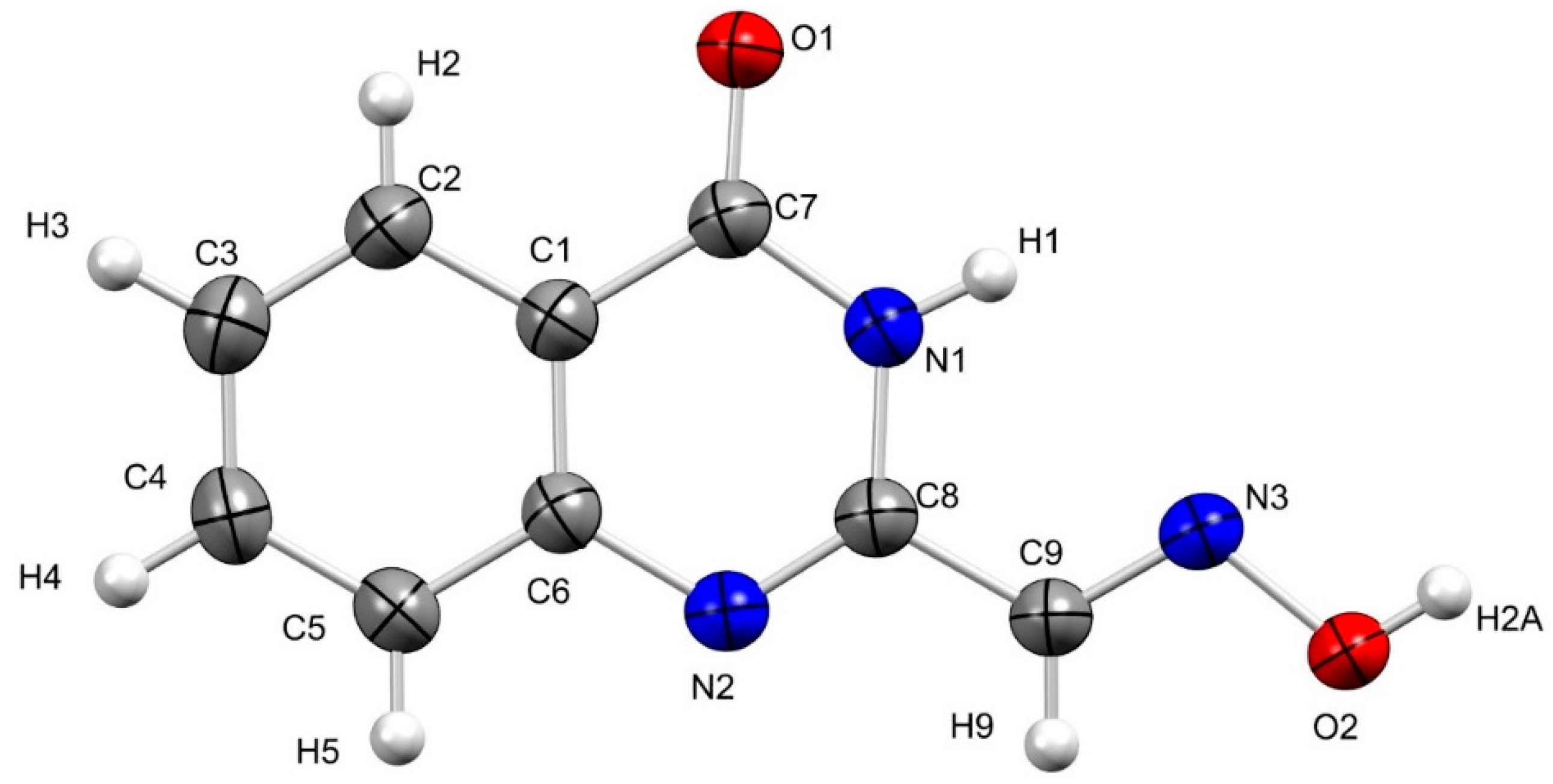
2. Results and Discussion
3. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Rewcastle, G.W. Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Ramsden, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Elsevier: Oxford, UK, 2008; Chapter 8.02; Volume 8, pp. 117–272. [Google Scholar]
- Kalogirou, A.S.; Kourtellaris, A.; Koutentis, P.A. Synthesis and Reactivity of 3′,5′-Dichloro-1H-spiro(quinazoline-2,4′-[1,2,6]thiadiazin)-4(3H)-ones. Eur. J. Org. Chem. 2019, 5462–5474. [Google Scholar] [CrossRef]
- Kalogirou, A.S.; Kourtellaris, A.; Koutentis, P.A. Synthesis of 2-Cyanoquinazolin-4-ones from 3′,5′-Dichloro-1H-spiro(quinazoline-2,4′-[1,2,6]thiadiazin)-4(3H)-ones. Chem. Select 2020, 5, 1884–1889. [Google Scholar] [CrossRef]
- Kalogirou, A.S.; Kourtellaris, A.; Koutentis, P.A. Synthesis and chemistry of benzo[e][1,2,6]thiadiazino[3,4-b][1,4]diazepin-10(11H)-ones and related ring transformations. J. Org. Chem. 2021, 86, 5702–5713. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Peng, B.; Xiao, B.B.; Cao, S.L.; Yang, C.R.; Wang, W.Z.; Wang, F.C.; Li, H.-Y.; Yuan, X.-L.; Shi, R.; et al. Synthesis and evaluation of chalcone analogues containing a 4-oxoquinazolin-2-yl group as potential anti-tumor agents. Eur. J. Med. Chem. 2019, 162, 586–601. [Google Scholar] [CrossRef] [PubMed]
- Neelakantan, L.; Hartung, W.H. α-Hydroxylamino Nitriles and α-Hydroxylamino Acids. J. Org. Chem. 1958, 23, 964–967. [Google Scholar] [CrossRef]
- Jin, C.; Burgess, J.P.; Kepler, J.A.; Cook, C.E. Copper-Catalyzed Cyclization of Steroidal Acylaminoacetylenes: Syntheses of Novel 11β-Aryl-17,17-spiro[(4′H,5′-methylene)oxazol]-Substituted Steroids. Org. Lett. 2007, 9, 1887–1890. [Google Scholar] [CrossRef] [PubMed]
- Maspero, M.; Volpato, D.; Cirillo, D.; Chen, N.Y.; Messerer, R.; Sotriffer, C.; De Amici, M.; Holzgrabe, U.; Dallanoce, C. Tacrine-xanomeline and tacrine-iperoxo hybrid ligands: Synthesis and biological evaluation at acetylcholinesterase and M1 muscarinic acetylcholine receptors. Bioorg. Chem. 2020, 96, 103633–103646. [Google Scholar] [CrossRef] [PubMed]
- Mamedov, V.A.; Zhukova, N.A.; Syakaev, V.V.; Gubaidullin, A.T.; Kadyrova, M.S.; Beschastnova, T.N.; Rizvanov, I.K.; Latypov, S.K. Simultaneous formation of 3-(benzimidazol-2-yl)quinoxalin-2(1H)-ones and 2-(benzimidazol-2-yl)quinoxalines from quinoxalin-2(1H)-one-3-carbaldoximes when exposed to 1,2-benzenediamines. Tetrahedron 2020, 76, 131721. [Google Scholar] [CrossRef]
- Kok, G.B.; Leung, B.K.Y.; Gautier, E.C.L.; Barnam, K.J. Neurologically Active Compounds. WO Patent 031161, 15 April 2004. [Google Scholar]
- Ginzinger, W.; Muhlgassner, G.; Arion, V.B.; Jakupec, M.A.; Roller, A.; Galanski, M.; Reithofer, M.; Berger, W.; Keppler, B.K. A SAR study of novel antiproliferative ruthenium and osmium complexes with quinoxalinone ligands in human cancer cell lines. J. Med. Chem. 2012, 55, 3398–3413. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalogirou, A.S.; Kourtellaris, A.; Koutentis, P.A. (E)-4-Oxo-3,4-dihydroquinazoline-2-carbaldehyde Oxime. Molbank 2021, 2021, M1233. https://doi.org/10.3390/M1233
Kalogirou AS, Kourtellaris A, Koutentis PA. (E)-4-Oxo-3,4-dihydroquinazoline-2-carbaldehyde Oxime. Molbank. 2021; 2021(2):M1233. https://doi.org/10.3390/M1233
Chicago/Turabian StyleKalogirou, Andreas S., Andreas Kourtellaris, and Panayiotis A. Koutentis. 2021. "(E)-4-Oxo-3,4-dihydroquinazoline-2-carbaldehyde Oxime" Molbank 2021, no. 2: M1233. https://doi.org/10.3390/M1233
APA StyleKalogirou, A. S., Kourtellaris, A., & Koutentis, P. A. (2021). (E)-4-Oxo-3,4-dihydroquinazoline-2-carbaldehyde Oxime. Molbank, 2021(2), M1233. https://doi.org/10.3390/M1233