
Unexpected Metal-Free Dehydrogenation of a β-Ketoester to a Phenol Using a Recyclable Oxoammonium Salt



Abstract
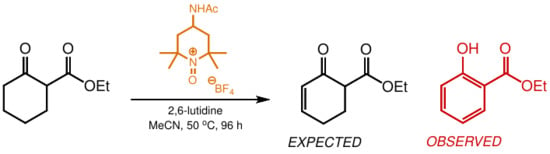
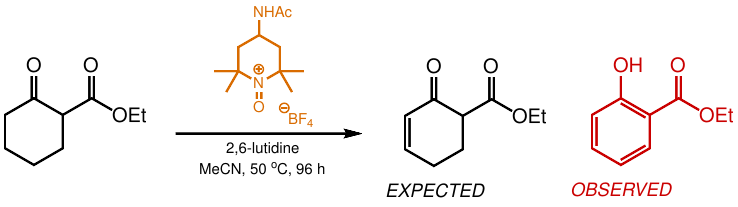
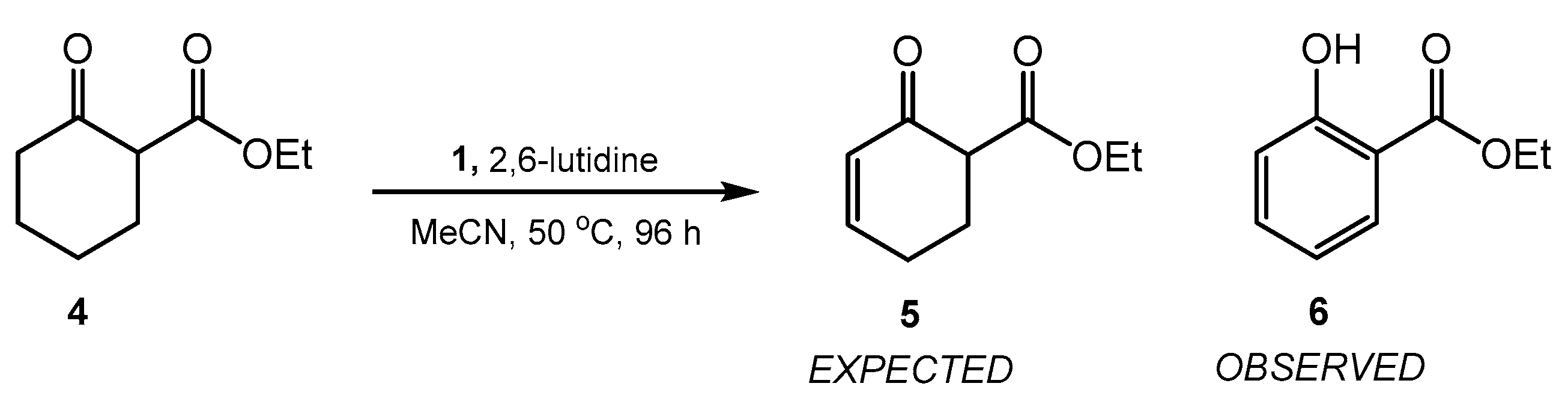
1. Introduction
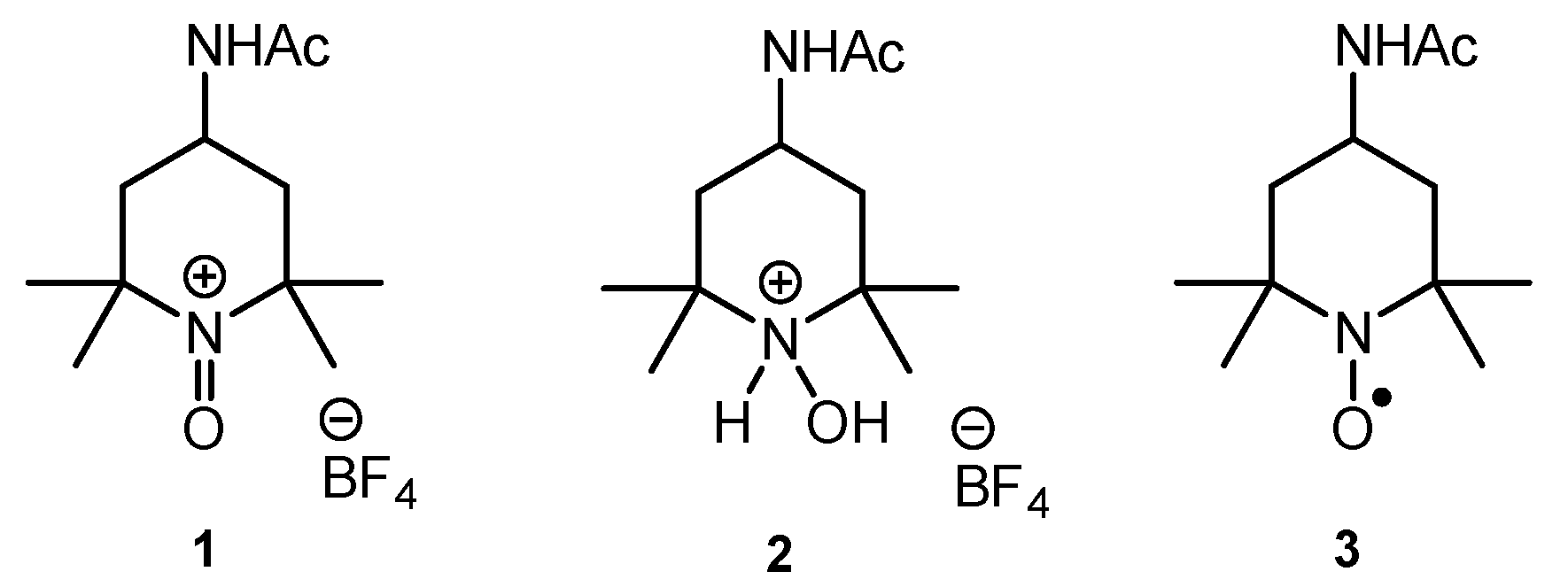
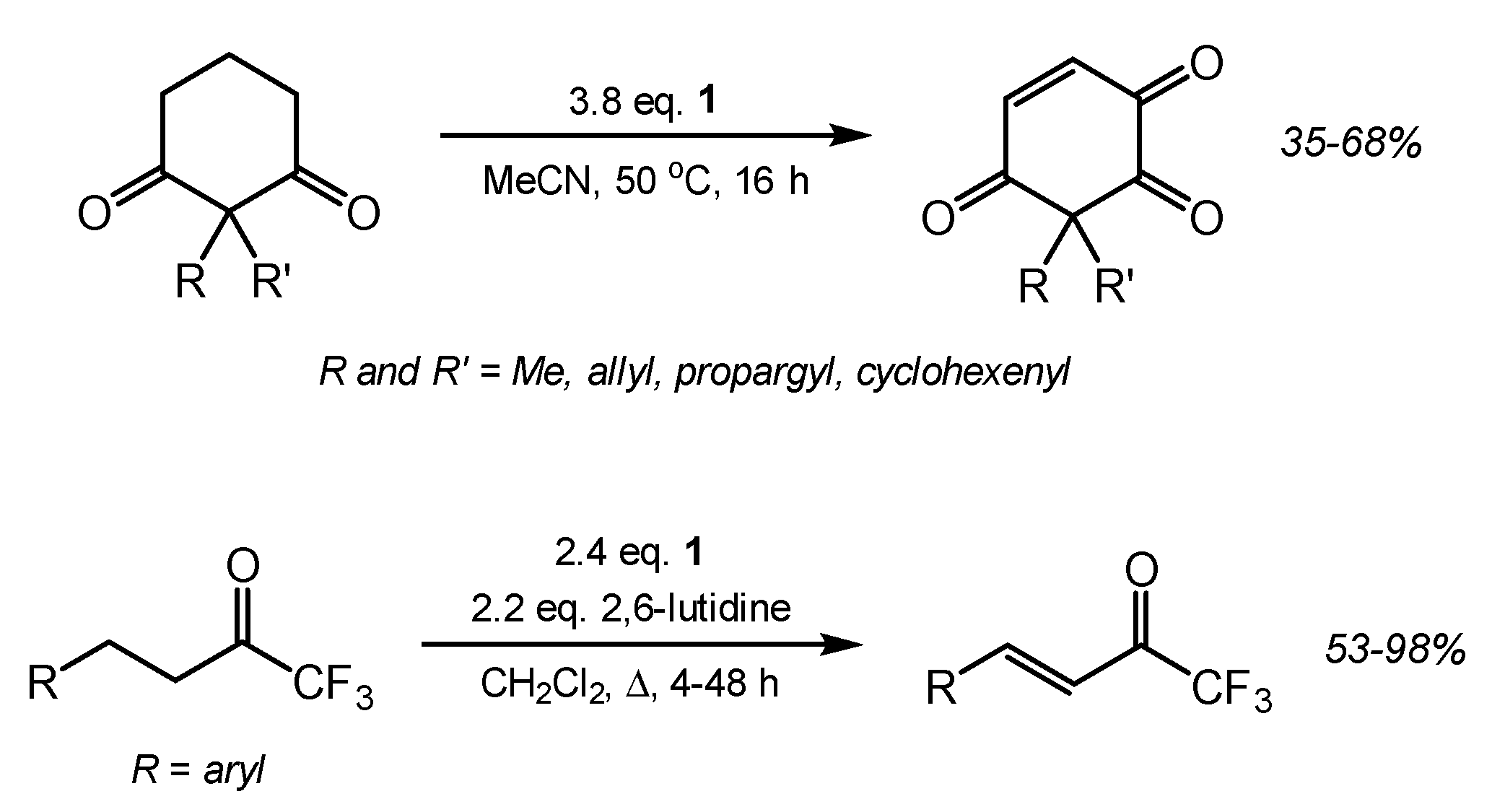
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Chemicals
3.3. Synthesis of Ethyl Salicylate (6) [CAS 118-61-6]
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shibuya, M. Nitroxyl Radical-Catalyzed Chemoselective Alcohol Oxidation for the Synthesis of Polyfunctional Molecules. Tetrahedron Lett. 2020, 61, 151515. [Google Scholar] [CrossRef]
- Beejapur, H.A.; Zhang, Q.; Hu, K.; Zhu, L.; Wang, J.; Ye, Z. TEMPO in Chemical Transformations: From Homogeneous to Heterogeneous. ACS Catal. 2019, 9, 2777–2830. [Google Scholar] [CrossRef]
- Tebben, L.; Studer, A. Nitroxides: Applications in synthesis and in polymer chemistry. Angew. Chem. Int. Ed. 2011, 50, 5034–5068. [Google Scholar] [CrossRef] [PubMed]
- Ciriminna, R.; Pagliaro, M. Industrial oxidations with organocatalyst TEMPO and its derivatives. Org. Proc. Res. Dev. 2010, 14, 245–251. [Google Scholar] [CrossRef]
- Bobbitt, J.M.; Brückner, C.; Merbouh, N. Oxoammonium-catalyzed oxidation. Org. React. 2009, 74, 103–206. [Google Scholar]
- Vogler, T.; Studer, A. Applications of TEMPO in Synthesis. Synthesis 2008, 1979–1993. [Google Scholar] [CrossRef]
- Kelly, C.B. 2,2,6,6-Tetramethylpiperidine-Based Oxoammonium Salts. Synlett 2013, 24, 527–528. [Google Scholar] [CrossRef]
- Leadbeater, N.E.; Bobbitt, J.M. TEMPO-Derived Oxoammonium Salts as Versatile Oxidizing Agents. Aldrichimica Acta 2014, 47, 65–74. [Google Scholar]
- Gini, A.; Brandhofer, T.; Mancheño, O.G. Recent Progress in Mild Csp3-H Bond Dehydrogenative or (Mono-) Oxidative Functionalization. Org. Biomol. Chem. 2017, 15, 1294–1312. [Google Scholar] [CrossRef]
- Garcia-Mancheño, O.; Stopka, T. TEMPO Derivatives as Alternative Mild Oxidants in Carbon–Carbon Coupling Reactions. Synthesis 2013, 45, 1602–1611. [Google Scholar] [CrossRef]
- Rohlmann, R.; Garcia-Mancheño, O. Metal-Free Oxidative C(sp3)-H Bond Couplings as Valuable Synthetic Tools for C–C Bond Formations. Synlett 2013, 24, 6–10. [Google Scholar] [CrossRef]
- Kelly, C.B.; Mercadante, M.A.; Wiles, R.J.; Leadbeater, N.E. Oxidative Esterification of Aldehydes Using a Recyclable Oxoammonium Salt. Org. Lett. 2013, 15, 2222–2225. [Google Scholar] [CrossRef] [PubMed]
- Ovian, J.M.; Kelly, C.B.; Pistritto, V.A.; Leadbeater, N.E. Accessing N-Acyl Azoles via Oxoammonium Salt-Mediated Oxidative Amidation. Org. Lett. 2017, 19, 1286–1289. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.B.; Lambert, K.M.; Mercadante, M.A.; Ovian, J.M.; Bailey, W.F.; Leadbeater, N.E. Access to Nitriles from Aldehydes Mediated by an Oxoammonium Salt. Angew. Chem. Int. Ed. 2015, 54, 4241–4245. [Google Scholar] [CrossRef]
- Twilton, J.; Le, C.; Zhang, P.; Shaw, M.H.; Evans, R.W.; MacMillan, D.W.C. The merger of transition metal and photocatalysis. Nat. Rev. Chem. 2017, 1, 52. [Google Scholar] [CrossRef]
- Shaw, M.H.; Twilton, J.; Macmillan, D.W.C. Photoredox Catalysis in Organic Chemistry. J. Org. Chem. 2016, 81, 6898–6926. [Google Scholar] [CrossRef]
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; Macmillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef]
- Nandi, J.; Vaughan, M.Z.; Sandoval, A.L.; Paolillo, J.M.; Leadbeater, N.E. Oxidative Amidation of Amines in Tandem with Transamidation: A Route to Amides Using Visible-Light Energy. J. Org. Chem. 2020, 85, 9219–9229. [Google Scholar] [CrossRef]
- Nandi, J.; Leadbeater, N.E. Visible-light-driven catalytic oxidation of aldehydes and alcohols to nitriles by 4-acetamido-tempo using ammonium carbamate as a nitrogen source. Org. Biomol. Chem. 2019, 17, 9182–9186. [Google Scholar] [CrossRef]
- Nandi, J.; Witko, M.L.; Leadbeater, N.E. Combining Oxoammonium Cation Mediated Oxidation and Photoredox Catalysis for the Conversion of Aldehydes into Nitriles. Synlett 2018, 29, 2185–2190. [Google Scholar]
- Pistritto, V.A.; Paolillo, J.M.; Bisset, K.A.; Leadbeater, N.E. Oxidation of α-trifluoromethyl and non-fluorinated alcohols via the merger of oxoammonium cations and photoredox catalysis. Org. Biomol. Chem. 2018, 16, 4715–4719. [Google Scholar] [CrossRef] [PubMed]
- Bosque, I.; Chinchilla, R.; Gonzalez-Gomez, J.C.; Guijarro, D.; Alonso, F. Cross-Dehydrogenative Coupling Involving Benzylic and Allylic C-H Bonds. Org. Chem. Front. 2020, 7, 1717–1742. [Google Scholar] [CrossRef]
- Bao, X.; Jiang, W.; Liang, J.; Huo, C. One-Electron Oxidative Dehydrogenative Annulation and Cyclization Reactions. Org. Chem. Front. 2020, 7, 2107–2144. [Google Scholar] [CrossRef]
- Eddy, N.A.; Kelly, C.B.; Mercadante, M.A.; Leadbeater, N.E.; Fenteany, G. Access to dienophilic ene-triketone synthons by oxidation of diketones with an oxoammonium salt. Org. Lett. 2012, 14, 498–501. [Google Scholar] [CrossRef]
- Hamlin, T.A.; Kelly, C.B.; Leadbeater, N.E. Dehydrogenation of Perfluoroalkyl Ketones Using a Recyclable Oxoammonium Salt. Eur. J. Org. Chem. 2013, 3658–3661. [Google Scholar] [CrossRef]
- Hamlin, T.A.; Kelly, C.B.; Ovian, J.M.; Wiles, R.J.; Tilley, L.J.; Leadbeater, N.E. Toward a Unified Mechanism for Oxoammonium Salt-Mediated Oxidation Reactions: A Theoretical and Experimental Study Using a Hydride Transfer Model. J. Org. Chem. 2015, 80, 8150–8167. [Google Scholar] [CrossRef]
- Bobbitt, J.M.; Bartelson, A.L.; Bailey, W.F.; Hamlin, T.A.; Kelly, C.B. Oxoammonium Salt Oxidations of Alcohols in the Presence of Pyridine Bases. J. Org. Chem. 2014, 79, 1055–1067. [Google Scholar] [CrossRef]
- Mercadante, M.; Kelly, C.B.; Bobbitt, J.M.; Tilley, L.J.; Leadbeater, N.E. Synthesis of 4-acetamido-2,2,6,6-tetramethylpiperidine-1-oxoammonium tetrafluoroborate and 4-acetamido-(2,2,6,6-tetramethyl-piperidin-1-yl)oxyl and their use in oxidative reactions. Nat. Protoc. 2013, 8, 666–676. [Google Scholar] [CrossRef]
- Cui, L.Q.; Dong, Z.L.; Liu, K.; Zhang, C. Design, Synthesis, Structure, and Dehydrogenation Reactivity of a Water Soluble o-Iodoxybenzoic Acid Derivative Bearing a Trimethylammonium Group. Org. Lett. 2011, 13, 6488–6491. [Google Scholar] [CrossRef]
- Samadi, S.; Orellana, A. A New Route to Phenols: Palladium-Catalyzed Cyclization and Oxidation of γ,δ-Unsaturated Ketones. ChemCatChem 2016, 8, 2472–2475. [Google Scholar] [CrossRef]
- Zhao, L.; Huang, G.; Guo, B.; Xu, L.; Chen, J.; Cao, W.; Zhao, G.; Wu, X. Diastereo- and Enantioselective Propargylation of Benzofuranones Catalyzed by Pybox-Copper Complex. Org. Lett. 2014, 16, 5584–5587. [Google Scholar] [CrossRef] [PubMed]
- Shome, S.; Singh, S.P. Design and synthesis of ruthenium bipyridine catalyst: An approach towards low-cost hydroxylation of arenes and heteroarenes. Tetrahedron Lett. 2017, 58, 3743–3746. [Google Scholar] [CrossRef]
- Bayguzina, A.R.; Tarisova, L.I.; Khusnutdinov, R.I. Synthesis of Hydroxybenzoic Acids and Their Esters by Reaction of Phenols with Carbon Tetrachloride and Alcohols in the Presence of Iron Catalysts. Russ. J. Gen. Chem. 2018, 88, 208–215. [Google Scholar] [CrossRef]
- Gopinath, R.; Barkakaty, B.; Talukdar, B.; Patel, B.K. Peroxovanadium-Catalyzed Oxidative Esterification of Aldehydes. J. Org. Chem. 2003, 68, 2944–2947. [Google Scholar] [CrossRef]
- Brenner, J.E. The Synthesis of 2-Carboethoxy-Δ2-cyclohexenone. J. Org. Chem. 1961, 26, 22–27. [Google Scholar] [CrossRef]
- Magano, J.; Chen, M.H.; Clark, J.D.; Nussbaumer, T. 2-(Diethylamino)ethanethiol, a New Reagent for the Odorless Deprotection of Aromatic Methyl Ethers. J. Org. Chem. 2006, 71, 7103–7105. [Google Scholar] [CrossRef]
- Parkins, M.V.; Kitching, W.; Drew, R.A.I.; Moore, C.J.; König, W.A. Chemistry of fruit flies: Composition of the male rectal gland secretions of some species of South-East Asian Dacinae. Re-examination of Dacus cucurbitae (melon fly). J. Chem. Soc. Perkin Trans. 1990, 1, 1111–1117. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | 1 (eq.) | 2,6-Lutidine (eq.) | Temperature (°C) | Time (h) | Conversion to 6 (%) b |
|---|---|---|---|---|---|
| 1 | 3.6 | 5 | 50 | 96 | 51 |
| 2 | 7.5 | 5 | 50 | 24 | 100 |
| 3 | 7.5 | 0 | 50 | 24 | 0 |
| 4 | 7.5 | 5 | 25 | 72 | 53 |
| 5 | 7.5 | 5 | 25 | 72 | 95 |
| 6 | 5.0 | 5 | 50 | 72 | 92 |
| 7 c | 7.5 | 5 | 100 | 0.5 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Politano, F.; Brydon, W.P.; Nandi, J.; Leadbeater, N.E. Unexpected Metal-Free Dehydrogenation of a β-Ketoester to a Phenol Using a Recyclable Oxoammonium Salt. Molbank 2021, 2021, M1180. https://doi.org/10.3390/M1180
Politano F, Brydon WP, Nandi J, Leadbeater NE. Unexpected Metal-Free Dehydrogenation of a β-Ketoester to a Phenol Using a Recyclable Oxoammonium Salt. Molbank. 2021; 2021(1):M1180. https://doi.org/10.3390/M1180
Chicago/Turabian StylePolitano, Fabrizio, William P. Brydon, Jyoti Nandi, and Nicholas E. Leadbeater. 2021. "Unexpected Metal-Free Dehydrogenation of a β-Ketoester to a Phenol Using a Recyclable Oxoammonium Salt" Molbank 2021, no. 1: M1180. https://doi.org/10.3390/M1180
APA StylePolitano, F., Brydon, W. P., Nandi, J., & Leadbeater, N. E. (2021). Unexpected Metal-Free Dehydrogenation of a β-Ketoester to a Phenol Using a Recyclable Oxoammonium Salt. Molbank, 2021(1), M1180. https://doi.org/10.3390/M1180