Abstract
γ-Glutamyl derivatives of sulfur amino acids have been prepared in multigram scale starting from readily available starting materials. The synthesis comprises two one-pot operations, both consisting of two reactions. In the first operation, N-phtaloyl-l-glutamic acid anhydride is obtained from l-glutamic acid and phtalic anhydride. In the second one, N-phtaloyl-l-glutamic acid anhydride is used to acylate amino acids and the N-phtaloyl protecting group is removed. The described approach offers a viable entry to γ-glutamyl derivatives of sulfur-containing amino acids with flavor-enhancer and nutraceutical properties.
1. Introduction
γ-Glutamyl derivatives are compounds in which the amino group of an amino acid or the N-terminal of a short peptide is acylated by the γ-carboxyl carbon of a glutamic acid residue. They are naturally occurring compounds; glutathione (γ-glutamylcysteinylglycine, GSH) is the most widespread thiol in cells, and various γ-glutamyl amino acids are common constituents of plants [1,2,3,4]. γ-Glutamyl derivatives of amino acids have altered chemico-physical and organoleptic properties, yet representing precursors of the parent compounds [5].
Several γ-glutamyl amino acids are taste-active compounds with kokumi properties found in many foods [6,7]. Kokumi is a Japanese word related to gustative sensations, usually described in terms of continuity, roundness, long-lasting savory sensations [8]. Kokumi compounds represent the ideal flavor enhancers, as they are able to reinforce the taste of food with threshold concentrations in the micromolar range [9]. Biological activities exerted by some γ-glutamyl derivatives are also known [10,11], and the fact that such compounds are constituents of food vegetables led to them being considered as nutraceuticals.
The applicative exploitation of the favorable characteristics of γ-glutamyl compounds is hampered by the difficulties connected with their availability in sufficient amount at a reasonable cost. Extraction from natural sources is laborious, low-yielding, and erratic, as it depends on the developmental stage of the plant and its conservation [12,13]. The (chemo) enzymatic synthesis of γ-glutamyl derivatives using bacterial γ-glutamyltransferases [14,15] or glutaminases [11], although considered as promising, still suffers from apparently unavoidable side reactions [16]. The synthesis through the classical peptide chemistry is plagued by the need for extensive use of protecting groups, complicated by the difficult differentiation of the two carboxyl groups in the glutamic acid moiety. As a consequence, a series of protection/deprotection steps involving orthogonal protecting groups are required in order to obtain a suitably protected glutamic acid with the free γ-carboxyl group. Thus, the desired γ-glutamyl derivatives are usually obtained in rather low yields after a number of steps [7].
In this context, the use of the cyclic anhydride of N-phtaloyl-l-glutamic acid (1) is advantageous for several reasons. In fact, this compound represents, on one side, an N-protected, and on the other, a carboxyl-activated glutamic acid moiety. Moreover, it has long since been known that cleavage of the anhydride ring leads regioselectively to γ-glutamyl derivatives [17].
Following our interest in taste-active compounds of natural origin [18], we report here a multigram scale conversion of the sulfur-containing amino acids S-methyl-l-cysteine (2), S-allyl-l-cysteine (3), S-benzyl-l-cysteine (4), and methionine (5) into the corresponding γ-glutamyl derivatives 8–11 through two one-pot operations, each involving two synthetic steps. In the first operation, N-phtaloyl-l-glutamic acid anhydride (1) was obtained starting from l-glutamic acid 6 and phtalic anhydride 7 [19]; in the second operation, the γ-glutamyl derivatives 8–11 were obtained through reaction of anhydride 1 with the proper amino acid 2–5, followed by one-pot removal of the N-phtaloyl protecting group.
2. Results and Discussion
Our aim was to find a relatively easy synthesis of γ-glutamyl derivatives in few steps starting from economical and readily accessible starting materials. Thus, although N-phtaloyl-l-glutamic acid and its anhydride 1 are both commercially available, they are rather expensive products and we decided to synthesize them from l-glutamic acid 6 and phtalic anhydride 7. This approach allowed us to gain a clearer picture of the whole synthetic plan starting from bulk chemicals.
Cyclic anhydride of N-phtaloyl-l-glutamic acid (1) was obtained through the method of Gu [19], with little modifications (Scheme 1). The reaction was carried out by heating equimolar amounts of l-glutamic acid 6 and phtalic anhydride 7 without solvent. The use of a Kugelrohr apparatus allowed the mixing of the mixture while heating and, at the same time, provided removal of the formed water by distillation. It is known that partial racemization of glutamic acid represents the major problem connected with the synthesis of its N-phtaloyl derivative [19,20,21]. Rigorous control of the temperature and the reaction time is of utmost importance in order to obtain satisfactory yields and to limit racemization. Better results were obtained by heating the reaction mixture to no more than 140 °C and avoiding prolonged heating after water distillation ceased from the melted mixture (10 min maximum). The cyclization of the intermediate N-phtaloyl-l-glutamic acid to afford the cyclic anhydride 1 proceeds in a one-pot fashion, dissolving the cooled melted mass in acetic anhydride and heating to 105 °C for a few minutes. It is worth noting that, under these conditions, the reaction is not complete and partial racemization cannot be avoided, but the enantiopure anhydride 1 is selectively recovered in satisfactory yield after crystallization upon addition of xylene [19]. In repeated runs, anhydride 1 was obtained in 52–55% yield.
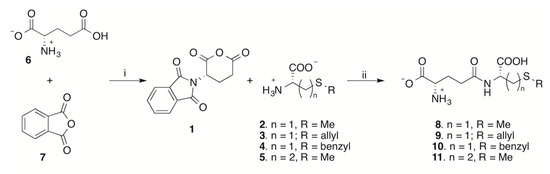
Scheme 1.
One-pot synthesis of γ-glutamyl derivatives 8–11. Reagents and conditions: (i) 140 °C with water removal by distillation, then Ac2O, 105 °C. (ii) DMF, 22 °C, then H2O, hydrazine hydrate 3.5 eq., precipitation with EtOH at 0 °C.
The acylation reaction of S-methylcysteine 2 with anhydride 1 was tried first. No protective group is required on the carboxyl group of the amino acid. The reaction was carried out in N,N-dimethylformamide (DMF), which proved to be the best solvent for the starting sulfur amino acid. Nevertheless, owing to the incomplete solubility of the starting amino acid, the reaction mixture was a suspension at the beginning and became a clear solution in time. It turned again into a cloudy suspension thanks to the low solubility of the reaction product. Raising the temperature up to 60 °C and/or the addition of little amounts of water to promote dissolution of the starting amino acid was revealed to be detrimental, resulting in the formation of a mixture of unidentified byproducts in the form of an intractable, viscous, sticky material. The addition of triethylamine as a base, in order to increase the nucleophilicity of the reacting amino group through deprotonation of the corresponding internal salt, resulted in an extensive racemization of the glutamic acid moiety. Thus, the reaction was carried out simply by mixing the two reactants in DMF at room temperature, regardless of the poor solubility of the starting amino acid. The N-phtaloyl-γ-glutamyl-S-methyl-l-cysteine 12 (Figure 1) was isolated as a foam after extractive aqueous work-up. The crude product proved to be suitable for complete 1- and 2D-NMR characterization, confirming its nature of γ-glutamyl compound (Figures S1–S4).
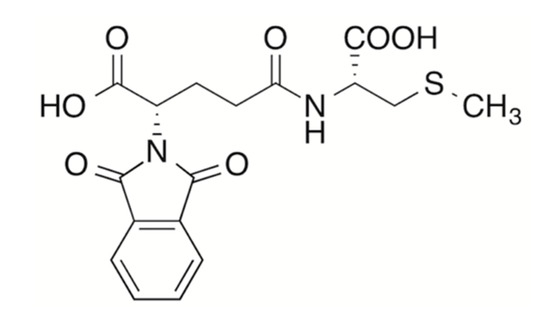
Figure 1.
N-phtaloyl-γ-glutamyl-S-methyl-l-cysteine 12.
The removal of the N-phtaloyl protecting group is usually obtained by reaction with hydrazine in alcoholic solvents [21,22]. We found that the reaction proceeds profitably in aqueous DMF in the presence of 3.5 equivalents of hydrazine hydrate. The amount of hydrazine hydrate is required by the presence of two free carboxyl groups in the molecule of the formed products. The product precipitates from the mixture as the hydrazinium salt, together with part of the byproduct phtaloyl hydrazide. Complete precipitation was promoted by rapid stirring at a low temperature after dilution with ethanol and the mixture of solids was recovered by filtration. Selective dissolution of the hydrazinium salt with water separated the desired γ-glutamyl derivative from the bulk of the less water-soluble phtaloyl-derived byproducts and ion exchange column chromatography afforded compound 8 in 93% isolated yield (50% from anhydride 1).
In order to ensure complete removal of hydrazine before elution of the γ-glutamyl derivatives from the ion exchange chromatographic column, the sensitive colorimetric assay based on the use of a 2% solution of N,N-dimethyl-4-aminobenzaldehyde in 10% aqueous HCl was employed (Figure S5) [23].
Having assessed the conditions for both the acylation reaction and for the removal of the N-phtaloyl protecting group, the two reactions were combined in a single one-pot operation. The nucleophilic amino acids 2–5 were used in slight excess (5 mol%) with respect to anhydride 1 in the acylation step. At the end of the acylation step, a little amount of water (10% with respect to the solvent DMF) is added to obtain complete dissolution of the suspended material and 3.5 equivalents hydrazine hydrate are used for N-phtaloyl group removal. Crude products were recovered by filtration as described. Compounds 8–11 were obtained in 60–98% yield from anhydride 1 after two synthetic steps and final ion exchange column chromatography.
3. Materials and Methods
3.1. General
l-glutamic acid, l-cysteine hydrochloride, S-methyl-l-cysteine, l-methionine, phtalic anhydride, hydrazine hydrate, and 4-(N,N-dimethylamino)benzaldehyde were from Sigma Aldrich (Darmstadt, Germany); S-allyl-l-cysteine and S-benzyl-l-cysteine were prepared by alkylation of l-cysteine hydrochloride with allyl bromide and benzyl bromide, respectively, according to the literature [24] and recrystallized from ethanol.
Analytical TLC was performed on silica gel F254 pre-coated aluminum sheets (0.2 mm layer) (Merck, Darmstadt, Germany) using nBuOH/water/acetic acid 4:1:1 as the eluent. Detection: UV lamp (λ 254 nm), 4.5% w/v CeSO4/(NH4)6Mo7O24·4H2O solution or 5% w/v ninhydrin solution in ethanol, followed by heating at 150 °C ca.
Ion exchange column chromatography was performed with Dowex 1 × 8 resin 200–400 mesh (Aldrich, Darmstadt, Germany) in the acetate form. Resin was equilibrated with 5 volumes of 0.5 M acetic acid before use.
1H-NMR and 13C-NMR spectra were acquired at 400.13 MHz and 100.61 MHz, respectively, on a Bruker Advance 400 spectrometer (Bruker, Karlsruhe, Germany) interfaced with a workstation running Windows operating system and equipped with a TOPSPIN software package. 13C signal multiplicities were based on attached proton test experiments (APT) and attributions were based on COSY (correlated spectroscopy), HSQC (hetero single quantum correlation), and HMBC (hetero multiple bond correlation) experiments. Chemical shifts are given in ppm (δ) and are referenced to solvent signals (δH CDCl3 7.26 ppm; δH D2O 4.79 ppm; δH DMSO 2.50 ppm; δC DMSO 39.52 ppm) or to TSP (3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt) as external standard (δMe 0.00 ppm). Spectra analyses were carried out with inmr Reader software (www.inmr.net).
Electrospray mass spectra (ESI-MS) were recorded on a Thermo Finnigan LCQ Advantage spectrometer (Hemel Hempstead, UK).
Melting points were measured with a Büchi Melting Point B-540 apparatus.
3.2. Synthesis of N-Phtaloyl-l-glutamic Acid Anhydride 1
l-glutamic acid 6 (4.44 g, 30.0 mmol) and phtalic anhydride 7 (4.41 g, 30.0 mmol) were mixed together and charged into a 50 mL round-bottom flask. The mixture of the two solids was heated to 140 °C while rotating the flask by means of a Kugelrohr apparatus. After melting, a pale yellow, oily mass was formed. The mass was cooled to 100 °C ca and acetic anhydride (10 mL) was added. The mixture was stirred in a pre-heated oil bath at 105 °C. After complete dissolution, heating continued for a further 10 min, and then the solution was transferred into a conical flask and xylene (15 mL) was added. The solution was allowed to slowly cool to room temperature, then the flask was closed and placed at 4 °C overnight. The formed colorless crystals were collected by filtration, washed in succession with small volumes of cold ethanol and diethyl ether, and dried under reduced pressure. Colorless crystals, 4.20 g, 54% yield.
1H-NMR (400 MHz; CDCl3): δ 7.89–7.93 (m, 2H), 7.82–7.77 (m, 2H), 5.10 (dd, J = 12.1, 5.9 Hz, 1H), 3.16 (dt, J = 17.1, 3.7 Hz, 1H), 2.97–2.78 (m, 2H), 2.22–2.15 (m, 1H).
[α = −42.1 (c 1.02, dioxane); lit. −44.7 (c 3, dioxane) [19].
3.3. Synthesis of N-Phtaloyl-γ-glutamyl-S-methyl-l-cysteine 12
To a well-stirred suspension of S-methyl-l-cysteine 2 (2.35 g, 17.36 mmol, 1.10 eq) in dry DMF (30 mL) at room temperature under a stream of dry nitrogen, N-phtaloyl-l-glutamic acid anhydride 1 was added in one portion (4.07 g, 15.70 mmol, 1.00 eq). At the end of the reaction (24 h, TLC monitoring), the mixture was diluted with water (200 mL), the pH was adjusted to ca 2 with 1 M HCl, and the resulting solution was extracted several times with AcOEt (80–100 mL portions, TLC monitoring). The combined organic layers were washed with saturated NaCl solution and dried over sodium sulfate, and the solvent was removed under reduced pressure. Colorless foam, 3.38 g, 53% yield. The product was characterized and used without further purification.
1H-NMR (400 MHz; DMSO-d6): δ 8.12 (d, J = 8.1 Hz, 1H, NH), 7.93–7.87 (m, 4H, aromatic H), 4.77 (dd, J = 10.6, 4.7 Hz, 1H, Hα Glu), 4.36 (td, J = 8.1, 5.5 Hz, 1H, Hα Cys), 2.77 (dd, J = 13.7, 5.5 Hz, 1H, Hβ1 Cys), 2.65 (dd, J = 13.7, 8.1 Hz, 1H, Hβ2 Cys), 2.43–2.32 and 2.32–2.24 (m, 1H each, Hβ Glu), 2.24–2.17 (m, 2H, Hγ Glu), 2.04 (s, 3H, S-Me).
13C-NMR (101 MHz, DMSO-d6): δ 15.19 (S-Me); 24.23 (Cβ Glu); 31.50 (Cγ Glu); 35.09 (Cβ Cys); 51.24 (Cα Glu); 51.40 (Cα Cys); 123.32 (aromatic CH); 131.25 (aromatic C); 134.74 (aromatic CH); 167.39 (phtalyl CO); 170.34 (α-COOH Glu); 171.05 (γ-COOH Glu); 172.11 (COOH Cys).
3.4. One-Pot Synthesis of γ-Glutamyl Derivatives of Sulfur Amino Acids.
3.4.1. General Procedure
The amino acid (15.75 mmol, 1.05 eq, 5 mol% excess with respect to the N-phtaloyl-l-glutamic acid anhydride) was suspended in dry DMF (30 mL, ca 2 mL/mmol of amino acid) under a stream of dry nitrogen. N-phtaloyl-l-glutamic acid anhydride 1 prepared as above (3.98 g, 15.00 mmol) was then added in one portion. At the completion of the reaction (ca 18–24 h, TLC monitoring), water was added (3 mL, 10% with respect to DMF), followed by hydrazine hydrate (3.5 eq with respect to the anhydride). A thick, white precipitate formed. The reaction is usually complete within 3–5 h (TLC monitoring). Ethanol was added (15 mL) and the suspension was vigorously stirred at 0 °C in an ice bath in order to favor complete precipitation. The solid was collected by filtration on a synthered glass septum and washed in succession with cold ethanol, ethyl acetate, and diethyl ether. The solid was then dried under reduced pressure without removing it from the septum. The hydrazinium salt of the desired product was removed from the septum by dissolving it with small aliquots of water, leaving the less soluble phtaloyl-derived byproducts on the septum. The pH of the solution was adjusted to ca 9.0 with 0.1 M NaOH and charged onto a pad of Dowex 1 × 8 ion exchange resin in the acetate form. The column was eluted with water until complete elution of hydrazine (test with 2% 4-(N,N-dimethylamino)benzaldehyde in 10% HCl) and then with 2 M acetic acid (three column volumes). The eluate was collected in fractions; fractions containing the desired product were combined on the basis of TLC analysis and freeze-dried. Compounds can be recrystallized from aqueous ethanol.
3.4.2. γ-Glutamyl-S-methyl-l-cysteine 8
Obtained from N-phtaloyl-l-glutamic acid anhydride 1 and S-methyl-l-cysteine 2 as a white solid, 3.05 g, 79% yield.
1H-NMR (400 MHz; DMSO-d6): δ 8.40 (d, J = 8.2 Hz, 1H, NH), 4.37 (ddd, J = 8.2, 8.2, 4.8 Hz, 1H, Hα Cys), 3.44 (t, J = 7.0 Hz, 1H, Hα Glu), 2.87 (dd, J = 13.6, 4.8 Hz, 1H, Hβ1 Cys), 2.73 (dd, J = 13.6, 8.2 Hz, 1H, Hβ2 Cys), 2.38–2.27 (m, 2H, Hγ Glu), 2.07 (s, 3H, S-Me), 1.82–1.90 (m, 1H, Hβ1 Glu); 1.90–1.98 (m, 1H, Hβ2 Glu).
13C-NMR (101 MHz; DMSO): δ 15.20 (S-Me), 26.95 (Cβ Glu), 31.58 (Cγ Glu), 35.25 (Cβ Cys), 52.00 (Cα Cys), 53.12 (Cα Glu), 170.41 (COOH Glu), 171.71 (CONH), 172.38 (COOH Cys).
ESI-MS, positive detection: m/z 265 [M + H]+; 529 [2M + H]+; 567 [2M + K]+; negative detection: m/z 263 [M − H]−.
m.p. = 104–106 °C (decomp.).
[α = −21.5 (c 1.03, H2O); lit. −17.9 (c 2.5, H2O) [25].
3.4.3. γ-Glutamyl-S-allyl-l-cysteine 9
Obtained from N-phtaloyl-l-glutamic acid anhydride 1 and S-allyl-l-cysteine 3 as a white solid, 3.45 g, 79% yield.
1H-NMR (400 MHz; DMSO-d6): δ 8.45 (d, J = 8.4 Hz, 1H, NH), 5.73 (ddt, J = 17.0, 9.9, 7.1 Hz, 1H, vinylc CH), 5.14–5.06 (m, 2H, vinylic CH2), 4.32 (ddd, J = 8.4, 8.4, 4.8 Hz, 1H, Hα Cys), 3.38 (t, J = 6.4 Hz, 1H, Hα Glu), 3.14 (d, J = 7.2 Hz, 2H, allylic CH2), 2.83 (dd, J = 13.6, 4.8 Hz, 1H, Hβ1 Cys), 2.66 (dd, J = 13.6, 8.4 Hz, 1H, Hβ2 Cys), 2.30 (t, J = 7.7 Hz, 2H, Hγ Glu), 1.98–1.81 (m, 2H, Hβ Glu).
13C-NMR (101 MHz; DMSO-d6): δ 27.08 (Cβ Glu); 31.67 (Cγ Glu); 31.76 (allylic CH2); 34.03 (Cβ Cys); 52.14 (Cα Cys); 53.21 (Cα Glu); 117.34 (vinylic CH2); 134.32 (vinylic CH); 170.17 (α-COOH Glu); 171.77 (CONH); 172.39 (COOH Cys).
ESI-MS, positive detection: m/z 291 [M + H]+; 313 [M + Na]+; negative detection: m/z 289 [M − H]−, 579 [2M − H]−.
m.p. = 155–157 °C (decomp.).
[α = −28.1 (c 1.03, H2O); lit. −17.0 (c 2.5, H2O) [25].
3.4.4. γ-Glutamyl-S-benzyl-l-cysteine 10
Obtained from N-phtaloyl-l-glutamic acid anhydride 1 and S-benzyl-l-cysteine 4 as a white solid, 3.00 g, 59% yield.
1H-NMR (400 MHz; DMSO-d6): δ 8.44 (d, J = 8.4 Hz, 1H, NH), 7.31–7.21 (m, 5H, aromatic H), 4.37 (ddd, J = 8.4, 8.4, 4.9 Hz, 1H, Hα Cys), 3.73 (s, 2H, benzylic CH2), 3.39 (t, J = 6.4 Hz, 1H, Hα Glu), 2.80 (dd, J = 13.6, 4.9 Hz, 1H, Hβ1 Cys), 2.65 (dd, J = 13.6, 8.4 Hz, 1H, Hβ2 Cys), 2.32 (t, J = 7.7 Hz, 2H, Hγ Glu), 2.01–1.83 (m, 2H, Hβ Glu).
13C-NMR (101 MHz; DMSO-d6): δ 27.55 (Cβ Glu); 32.22 (Cγ Glu); 33.02 (Cβ Cys); 35.82 (benzylic CH2); 52.62 (Cα Cys); 53.88 (Cα Glu); 127.30, 128.76 and 130.46 (aromatic CH); 138.76 (aromatic C); 171.37 (COOH Glu); 172.90 (CONH); 173.48 (COOH Cys).
ESI-MS, positive detection: m/z 341 [M + H]+, 385 [M – H + 2Na]+.
m.p. = 180–183 °C (decomp.).
[α = −14.6 (c 0.20, H2O); lit. −14.7 (c 0.03, H2O) [4].
3.4.5. γ-Glutamyl-l-methionine 11
Obtained from N-phtaloyl-l-glutamic acid anhydride 1 and l-methionine 5 as a white solid, 4.11 g, 98% yield.
1H-NMR (400 MHz; DMSO-d6): δ 8.43 (d, J = 7.7 Hz, 1H, NH), 4.27 (ddd, J = 8.4, 8.4, 4.2 Hz, 1H, Hα Met), 3.41 (t, J = 6.3 Hz, 1H, Hα Glu), 2.45–2.50 (m, 2H, Hγ Met), 2.31 (t, J = 7.7 Hz, 2H, Hγ Glu), 2.04 (s, 3H, S-Me), 1.98–1.82 (m, 4H, Hβ Glu and Hβ Met).
13C-NMR (101 MHz; DMSO-d6): δ 14.55 (S-Me); 26.94 (Cβ Glu); 29.75 (Cγ Met); 30.79 (Cβ Met); 31.52 (Cγ Glu); 51.37 (Cα Met); 53.16 (Cα Glu); 170.38 (COOH Glu); 171.92 (CONH); 173.54 (COOH Met).
ESI-MS, negative detection: m/z 277 [M − H]−, 299 [M − 2H + Na]−, 621 [2M − 4H + 3Na]−.
m.p. = 187–190 °C (decomp.).
[α = −9.7 (c 1.03, H2O]; lit. −9.2 (c 2.5, H2O) [25].
4. Conclusions
A simple, straightforward synthesis of γ-glutamyl derivatives of sulfur-containing amino acids was accomplished through a two one-step approach based on the use of N-phtaloyl-l-glutamic acid anhydride. No protecting groups on the nucleophilic amino acids are needed. The reaction rate and yield of the desired compounds are related to the solubility of the starting amino acid, with better yields and shorter reaction times being achieved with readily soluble amino acids. Our method provides the desired compounds in a few simple steps, as well as good yields and with practically complete optical purity starting from cheap, bulk starting materials.
Supplementary Materials
The following are available online, Supplementary material part 1: Complete characterization (1D- and 2D-NMR spectra and ESI mass spectra) of compounds 8–11; Supplementary material part 2, Figures S1–S4: 1D- and 2D-NMR spectra of compound 12; Figure S5: colorimetric assay for the removal of hydrazine by ion exchange column chromatography.
Author Contributions
Investigation, M.R. and N.V.; writing—review and editing, G.S.; supervision and writing—original draft, C.F.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was carried out within the TailGluTran Project, funded by Fondazione Cariplo, grant number 2016-0741.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kasai, T.; Shiroshita, Y.; Sakamura, S. gamma-Glutamyl peptides of Vigna radiata seeds. Phytochemistry 2013, 25, 679–682. [Google Scholar] [CrossRef]
- Muetsch-Eckner, M.; Meier, B.; Wright, A.D.; Sticher, O. γ-Glutamyl peptides from Allium sativum bulbs. Phytochemistry 1992, 31, 2389–2391. [Google Scholar] [CrossRef]
- Giada, M.D.L.R.; Miranda, M.T.M.; Marquez, U.M.L. Sulfur γ-glutamyl peptides in mature seeds of common beans (Phaseolus vulgaris L.). Food Chem. 1998, 61, 177–184. [Google Scholar] [CrossRef]
- Kubec, R.; Musah, R.A. γ-Glutamyl dipeptides in Petiveria alliacea. Phytochemistry 2005, 66, 2494–2497. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Lisk, D.; Block, E.; Ip, C. Characterization of the biological activity of γ-glutamyl-Se-methylselenocysteine. Cancer Res. 2001, 61, 2923–2928. [Google Scholar] [PubMed]
- Zhao, C.J.; Schieber, A.; Gänzle, M.G. Formation of taste-active amino acids, amino acid derivatives and peptides in food fermentations—A review. Food Res. Int. 2016, 89, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Amino, Y.; Wakabayashi, H.; Akashi, S.; Ishiwatari, Y. Structural analysis and taste evaluation of γ-glutamyl peptides comprising sulfur-containing amino acids. Biosci. Biotechnol. Biochem. 2018, 82, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, Y.; Yasuda, R.; Kuroda, M.; Eto, Y. Kokumi substances, enhancers of basic tastes, induce responses in calcium-sensing receptor expressing taste cells. PLoS ONE 2012, 7, e34489. [Google Scholar] [CrossRef]
- Yang, J.; Bai, W.; Zeng, X.; Cui, C. g-Glutamyl peptides: The food source, enzymatic synthesis, kokumi-active and the potential functional properties—A review. Trends Food Sci. Technol. 2019, 91, 339–346. [Google Scholar] [CrossRef]
- Wetli, H.A.; Brenneisen, R.; Tschudi, I.; Langos, M.; Bigler, P.; Sprang, T.; Schuerch, S.; Muehlbauer, R.C. A γ-Glutamyl peptide isolated from onion (Allium cepa L.) by bioassay-guided fractionation inhibits resorption activity of osteoclasts. J. Agric. Food Chem. 2005, 53, 3408–3414. [Google Scholar] [CrossRef]
- Yang, J.; Sun, D.; Cui, C.; Dong, K.; Zhao, M. γ-Glu-Met synthesised using a bacterial glutaminase as a potential inhibitor of dipeptidyl peptidase IV. Int. J. Food Sci. Technol. 2018, 53, 1166–1175. [Google Scholar] [CrossRef]
- Ichikawa, M.; Ide, N.; Ono, K. Changes in organosulfur compounds in garlic cloves during storage. J. Agric. Food Chem. 2006, 54, 4849–4854. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Hu, D.; Jiang, Y.; Chen, F.; Hu, X.; Zhao, G. Relationship between γ-glutamyl transpeptidase activity and garlic greening, as controlled by temperature. J. Agric. Food Chem. 2008, 56, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Yamada, C.; Kato, K. γ-Glutamyl compounds and their enzymatic production using bacterial γ-glutamyltranspeptidase. Amino Acids 2007, 32, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-Y.; Lo, H.-F.; Wang, T.-F.; Lin, M.-G.; Lin, L.-L.; Chi, M.-C. Enzymatic synthesis of γ-l-glutamyl-S-allyl-l-cysteine, a naturally occurring organosulfur compound from garlic, by Bacillus licheniformis γ-glutamyltranspeptidase. Enzyme Microb. Technol. 2015, 75, 18–24. [Google Scholar] [CrossRef]
- Morelli, C.F.; Calvio, C.; Biagiotti, M.; Speranza, G. pH-Dependent hydrolase, glutaminase, transpeptidase and autotranspeptidase activities of Bacillus subtilis γ-glutamyltransferase. FEBS J. 2014, 281, 232–245. [Google Scholar] [CrossRef]
- Sheehan, J.C.; Bolhofer, W.A. The structure of hydroxylysine. J. Am. Chem. Soc. 1950, 72, 2469–2472. [Google Scholar] [CrossRef]
- Speranza, G.; Morelli, C.F. γ-Glutamyl transpeptidase-catalyzed synthesis of naturally occurring flavor enhancers. J. Mol. Catal. B Enzym. 2012, 84, 65–71. [Google Scholar] [CrossRef]
- Gu, H.; Jiang, Y. A practical preparation of N,N-Phthalyl-l-glutamic 1,5-anhydride. Org. Prep. Proc. Int. 2004, 36, 479–481. [Google Scholar] [CrossRef]
- Tipson, S.R. N,N-Phtaloyl-l-glutamic acid and some derivatives. J. Org. Chem. 1956, 21, 1353–1356. [Google Scholar] [CrossRef]
- Magnan, S.D.J.; Shirota, F.N.; Nagasawa, H.T. Drug latentiation by gamma-glutamyl transpeptidase. J. Med. Chem. 1982, 25, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Menard, A.; Castonguay, R.; Lherbet, C.; Rivard, C.; Roupioz, Y.; Keillor, J.W. Nonlinear free energy relationship in the general-acid-catalyzed acylation of rat kidney γ-glutamyl transpeptidase by a series of γ-glutamyl anilide substrate analogues. Biochemistry 2001, 40, 12678–12685. [Google Scholar] [CrossRef] [PubMed]
- Feigl, F. Detection of characteristic functional groups in organic compounds. In Spots Tests in Organic Analysis, 7th ed.; Elsevier Publishing Company: Amsterdam, The Netherlands, 1966; pp. 277–279. [Google Scholar]
- Goudreau, N.; Brochu, C.; Cameron, D.R.; Duceppe, J.-S.; Faucher, A.-M.; Ferland, J.-M.; Grand-Maître, C.; Poirier, M.; Simoneau, B.; Tsantrizos, Y.S. Potent inhibitors of the hepatitis C virus NS3 protease: Design and synthesis of macrocyclic substrate-based β-strand mimics. J. Org. Chem. 2004, 69, 6185–6201. [Google Scholar] [CrossRef] [PubMed]
- Carson, J.F.; Wong, F.F. Preparation of γ-glutamyl dipeptides of sulfur-containing amino-acids. J. Chem. Soc. Perkin Trans. 1974, 1, 685–687. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).