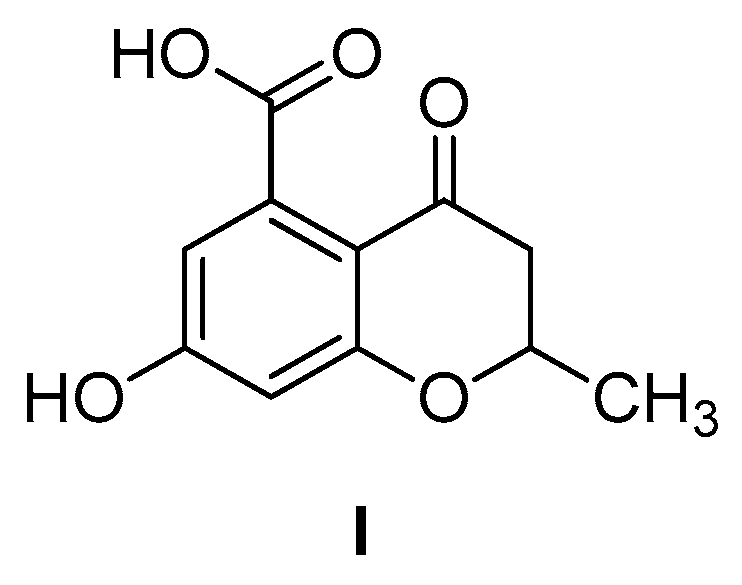
6-Hydroxy-2-methylbenzofuran-4-carboxylic Acid

 , ,
, ,  ,
, 
 and
and
Abstract
1. Introduction
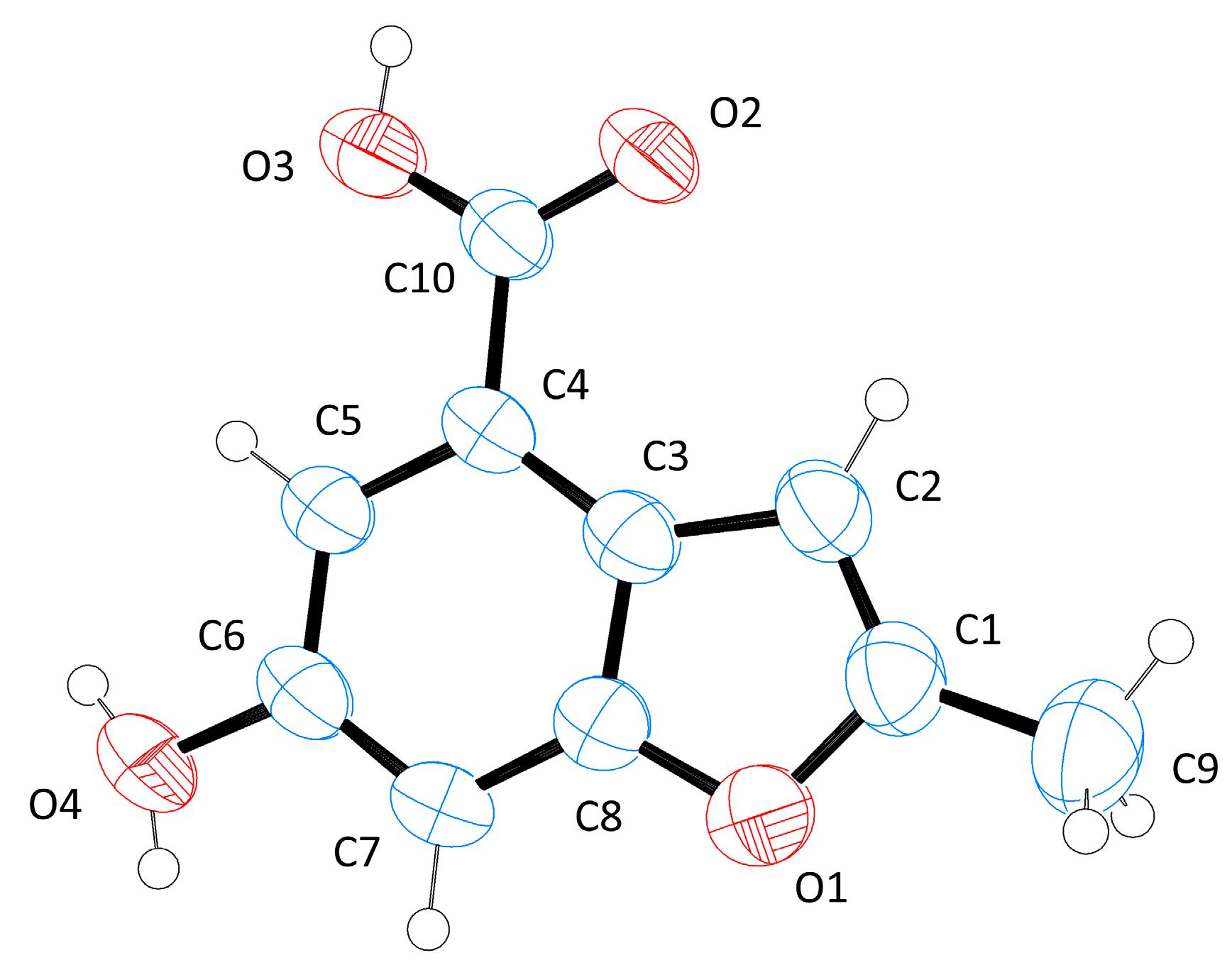
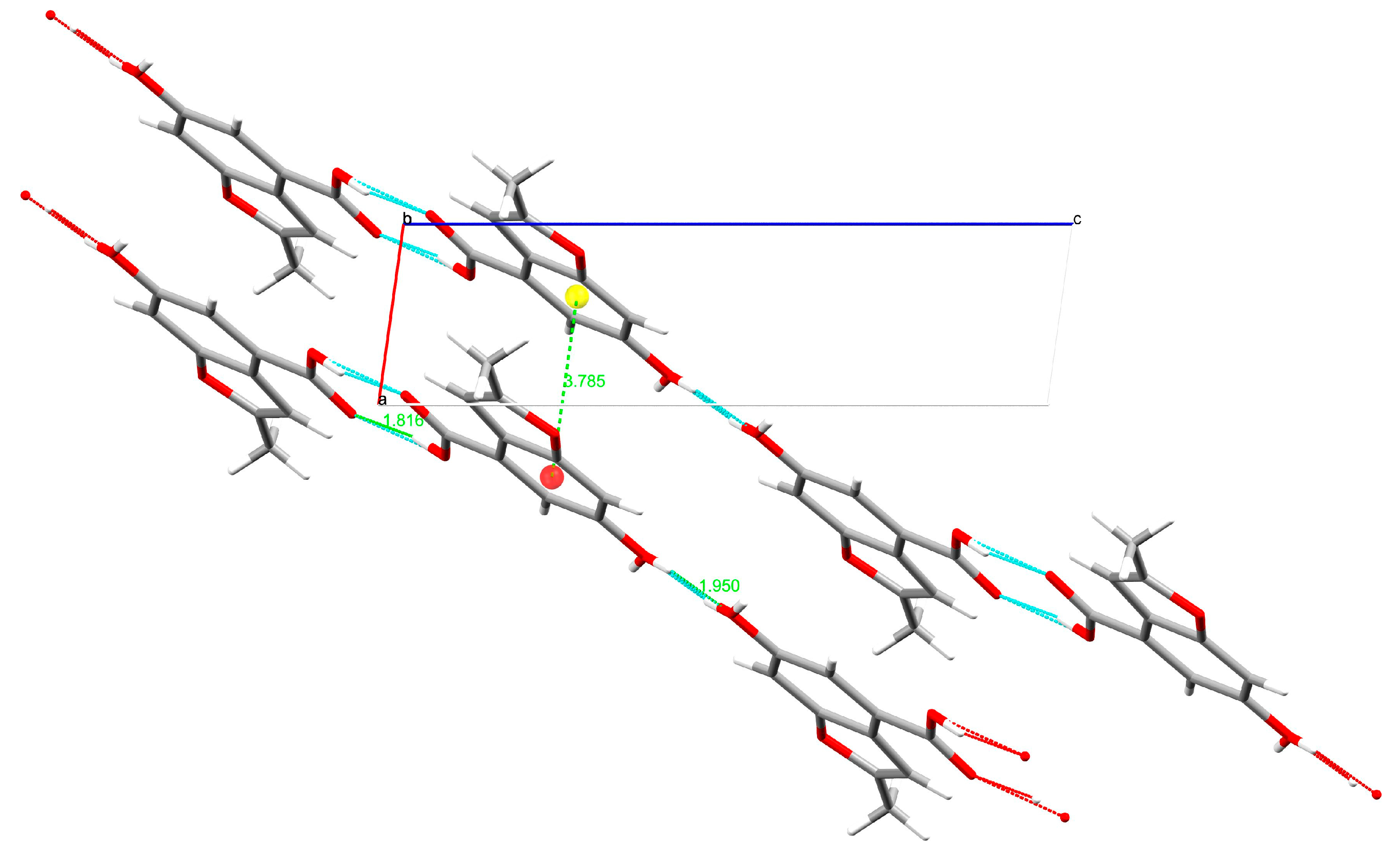
2. Results and Discussion
3. Materials and Methods
3.1. Synthesis
3.1.1. 6-Hydroxy-2-methylbenzofuran-4-carboxylic Acid Methyl Ester (2)
3.1.2. 6-Hydroxy-2-methylbenzofuran-4-carboxylic Acid (1)
3.2. Single-Crystal X-ray Diffraction
3.3. Enzymatic Assays
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Global Tuberculosis Report 2019; World Health Organization: Geneva, Switzerland, 2019; ISBN 9789241565646.
- Sotgiu, G.; Centis, R.; D’Ambrosio, L.; Migliori, G.B. Tuberculosis treatment and drug regimens. Cold Spring Harb. Perspect. Med. 2015, 5, a017822. [Google Scholar] [CrossRef] [PubMed]
- Chiarelli, L.R.; Mori, G.; Esposito, M.; Orena, B.S.; Pasca, M.R. New and old hot drug targets in tuberculosis. Curr. Med. Chem. 2016, 23, 3813–3846. [Google Scholar] [CrossRef]
- Fanzani, L.; Porta, F.; Meneghetti, F.; Villa, S.; Gelain, A.; Lucarelli, A.P.; Parisini, E. Mycobacterium tuberculosis low molecular weight phosphatases (MPtpA and MPtpB): From biological insight to inhibitors. Curr. Med. Chem. 2015, 22, 3110–3132. [Google Scholar] [CrossRef]
- Mori, M.; Sammartino, J.C.; Costantino, L.; Gelain, A.; Meneghetti, F.; Villa, S.; Chiarelli, L.R. An overview on the potential antimycobacterial agents targeting serine/threonine protein kinases from Mycobacterium tuberculosis. Curr. Top. Med. Chem. 2019, 19, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Truzzi, E.; Meneghetti, F.; Mori, M.; Costantino, L.; Iannuccelli, V.; Maretti, E.; Domenici, F.; Castellano, C.; Rogers, S.; Capocefalo, A.; et al. Drugs/lamellae interface influences the inner structure of double-loaded liposomes for inhaled anti-TB therapy: An in-depth small-angle neutron scattering investigation. J. Colloid Interface Sci. 2019, 541, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Meneghetti, F.; Villa, S.; Gelain, A.; Barlocco, D.; Chiarelli, L.R.; Pasca, M.R.; Costantino, L. Iron acquisition pathways as targets for antitubercular drugs. Curr. Med. Chem. 2016, 23, 4009–4026. [Google Scholar] [CrossRef]
- Chiarelli, L.R.; Mori, M.; Barlocco, D.; Beretta, G.; Gelain, A.; Pini, E.; Porcino, M.; Mori, G.; Stelitano, G.; Costantino, L.; et al. Discovery and development of novel salicylate synthase (MbtI) furanic inhibitors as antitubercular agents. Eur. J. Med. Chem. 2018, 155, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Chiarelli, L.R.; Mori, M.; Beretta, G.; Gelain, A.; Pini, E.; Sammartino, J.C.; Stelitano, G.; Barlocco, D.; Costantino, L.; Lapillo, M.; et al. New insight into structure-activity of furan-based salicylate synthase (MbtI) inhibitors as potential antitubercular agents. J. Enzyme Inhib. Med. Chem. 2019, 34, 823–828. [Google Scholar] [CrossRef]
- Mori, M.; Stelitano, G.; Gelain, A.; Pini, E.; Chiarelli, L.R.; Sammartino, J.C.; Poli, G.; Tuccinardi, T.; Beretta, G.; Porta, A.; et al. Shedding X-ray light on the role of magnesium in the activity of M. tuberculosis salicylate synthase (MbtI) for drug design. J. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Pini, E.; Poli, G.; Tuccinardi, T.; Chiarelli, L.; Mori, M.; Gelain, A.; Costantino, L.; Villa, S.; Meneghetti, F.; Barlocco, D. New chromane-based derivatives as inhibitors of Mycobacterium tuberculosis Salicylate Synthase (MbtI): Preliminary biological evaluation and molecular modeling studies. Molecules 2018, 23, 1506. [Google Scholar] [CrossRef]
- Palisse, A.; Kirsch, S.F. Synthesis of furans through silver-catalyzed propargyl-Claisen rearrangement followed by cyclocondensation. Eur. J. Org. Chem. 2014, 2014, 7095–7098. [Google Scholar] [CrossRef]
- More, K.R. Review on synthetic routes for synthesis of benzofuran-based compounds. J. Chem. Pharm. Res. 2017, 9, 210–220. [Google Scholar]
- Farrugia, L.J. ORTEP-3 for Windows—A version of ORTEP-III with a graphical user interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]
- Nardelli, M. PARST 95—An update to PARST: A system of Fortran routines for calculating molecular structure parameters from the results of crystal structure analyses. J. Appl. Crystallogr. 1995, 28, 659. [Google Scholar] [CrossRef]
- SMART & SAINT Software Reference Manual; Version 6.45; Bruker AXS, Inc.: Madison, WI, USA, 2003.
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement via SIR2014. J. Appl. Crystallogr. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 9–18. [Google Scholar] [CrossRef]
- MacRae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Vasan, M.; Neres, J.; Williams, J.; Wilson, D.J.; Teitelbaum, A.M.; Remmel, R.P.; Aldrich, C.C. Inhibitors of the Salicylate Synthase (MbtI) from Mycobacterium tuberculosis Discovered by High-Throughput Screening. ChemMedChem 2010, 5, 2079–2087. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H-Bond | H∙∙∙A (Å) | D∙∙∙A (Å) | D-H∙∙∙A (°) |
|---|---|---|---|
| O3-H3∙∙∙O2 | 1.82(1) | 2.623(1) | 167.8(1) |
| O4-H42∙∙∙O4 | 1.95(1) | 2.745(1) | 165.4(1) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mori, M.; Meneghetti, F.; Chiarelli, L.R.; Diego, A.; Nava, D.; Gelain, A.; Cazzaniga, G.; Villa, S.; Pini, E. 6-Hydroxy-2-methylbenzofuran-4-carboxylic Acid. Molbank 2020, 2020, M1143. https://doi.org/10.3390/M1143
Mori M, Meneghetti F, Chiarelli LR, Diego A, Nava D, Gelain A, Cazzaniga G, Villa S, Pini E. 6-Hydroxy-2-methylbenzofuran-4-carboxylic Acid. Molbank. 2020; 2020(2):M1143. https://doi.org/10.3390/M1143
Chicago/Turabian StyleMori, Matteo, Fiorella Meneghetti, Laurent R. Chiarelli, Alessia Diego, Donatella Nava, Arianna Gelain, Giulia Cazzaniga, Stefania Villa, and Elena Pini. 2020. "6-Hydroxy-2-methylbenzofuran-4-carboxylic Acid" Molbank 2020, no. 2: M1143. https://doi.org/10.3390/M1143
APA StyleMori, M., Meneghetti, F., Chiarelli, L. R., Diego, A., Nava, D., Gelain, A., Cazzaniga, G., Villa, S., & Pini, E. (2020). 6-Hydroxy-2-methylbenzofuran-4-carboxylic Acid. Molbank, 2020(2), M1143. https://doi.org/10.3390/M1143