Synthesis of N-Heterocyclic Analogues of 28-O-Methyl Betulinate, and Their Antibacterial and Antifungal Properties



Abstract

1. Introduction
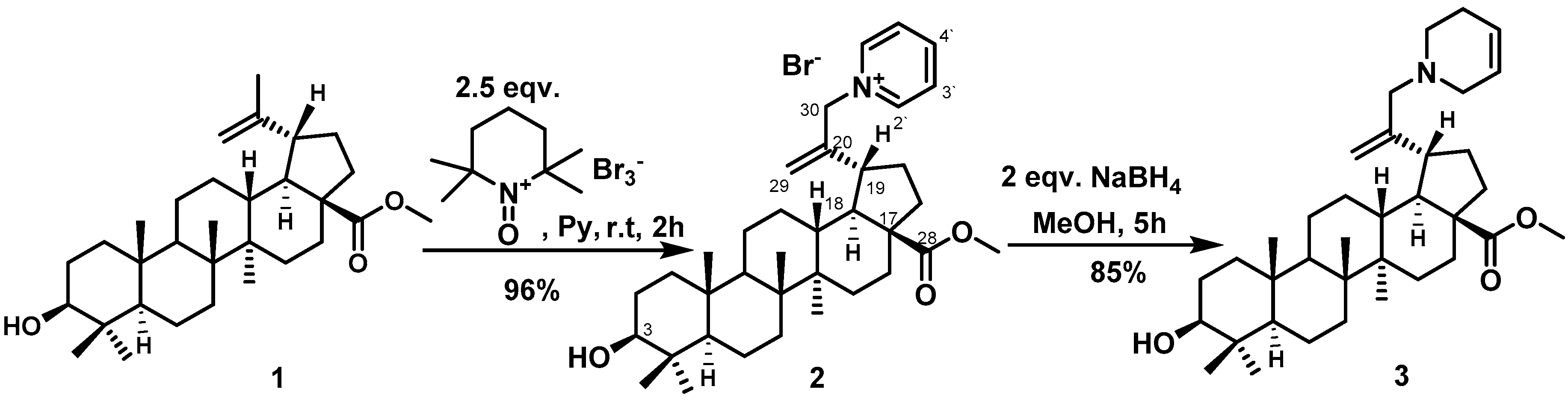
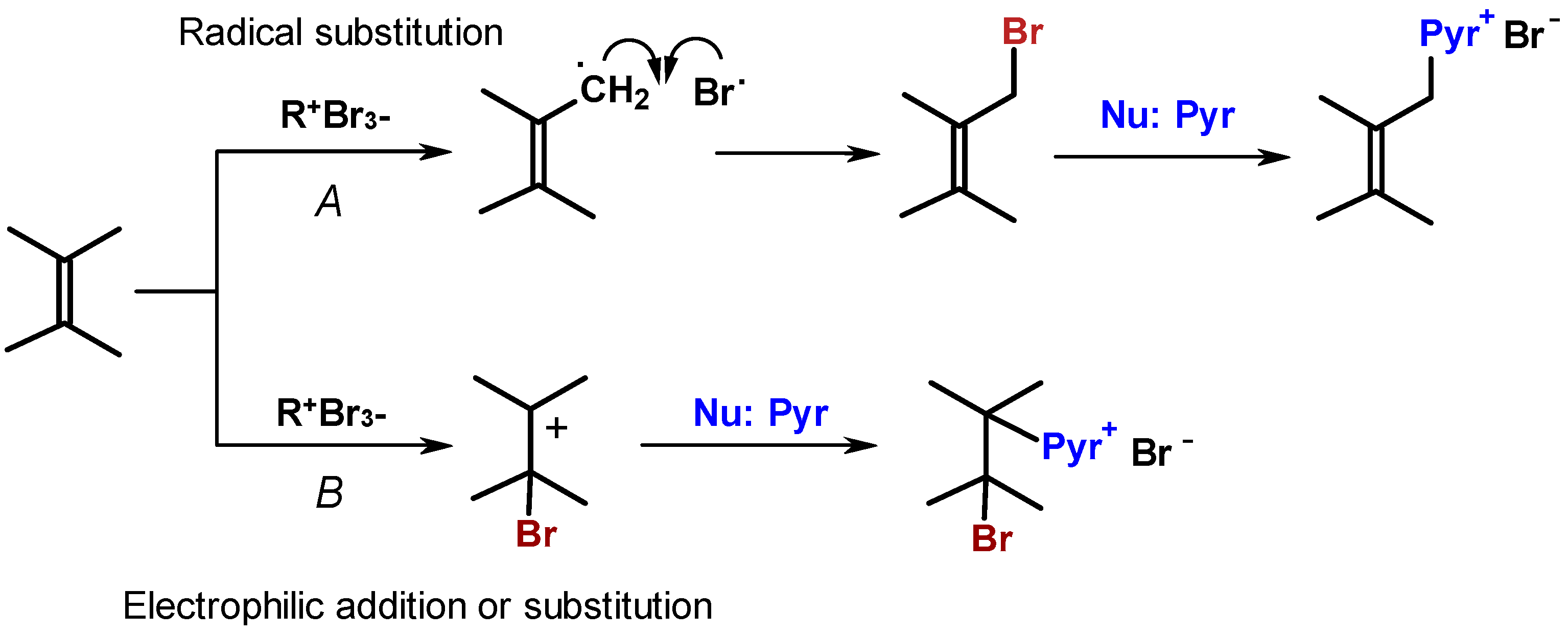
2. Results and Discussion
3. Materials and Methods
3.1. 1-{2-[(1R, 3aS, 5aR, 5bR, 7aR, 9S, 11aR, 13aR, 13bS)-9-hydroxy-3a-(methoxycarbonyl)-5a, 5b, 8, 8, 11a-pentamethyl-icosahydro-1H-cyclopenta [a] chrysen-1-yl] prop-2-en-1-yl} pyridin-1-ium bromide (2)
3.2. Methyl (1R, 3aS, 5aR, 5bR, 7aR, 9S, 11aR, 13aR, 13bS)-9-hydroxy-5a, 5b, 8, 8, 11a-pentamethyl-1-[3-(1,2,5,6-tetrahydropyridin-1-yl)prop-1-en-2-yl]-icosahydro-1H-cyclopenta [a] chrysene 3a-carboxylate (3)
3.3. Biological Screening
3.3.1. Antimicrobial Assay
3.3.2. Antifungal Assay
3.3.3. Hit Confirmation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Asif, M. Biological significances of nitrogen hetero atom containing heterocyclic compounds. Int. J. Bioorg. Chem. 2017, 2, 146–152. [Google Scholar] [CrossRef]
- Sundararaman, M.; Kumar, R.R.; Venkatesan, P.; Ilangovan, A. 1-Alkyl-(N,N-dimethylamino)pyridinium bromides: inhibitory effect on virulence factors of Candida albicans and on the growth of bacterial pathogens. J. Med. Microbiol. 2013, 62, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Serafim, T.L.; Carvalho, F.S.; Bernardo, T.C.; Pereira, G.C.; Perkins, E.; Holy, J.; Krasutsky, D.A.; Kolomitsyna, O.N.; Krasutsky, P.A.; Oliveira, P.J. New derivatives of lupane triterpenoids disturb breast cancer mitochondria and induce cell death. Bioorg. Med. Chem. 2014, 22, 6270–6287. [Google Scholar] [CrossRef]
- Alakurtti, S.; Mäkelä, T.; Koskimies, S.; Yli-Kauhaluoma, J. Pharmacological properties of the ubiquitous natural product betulin. Eur. J. Pharm. Sci. 2006, 29, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Furtado, N.A.J.C.; Pirson, L.; Edelberg, H.; Miranda, L.M.; Loira-Pastoriza, C.; Preat, V.; Larondelle, Y.; André, C.M. Pentacyclic triterpene bioavailability: An overview of in vitro and in vivo studies. Molecules 2017, 22, 400–424. [Google Scholar] [CrossRef] [PubMed]
- Holy, J.; Kolomitsyna, O.; Krasutsky, D.; Oliveira, P.J.; Perkins, E.; Krasutsky, P.A. Dimethylaminopyridine derivatives of lupane triterpenoids are potent disruptors of mitochondrial structure and function. Bioorg. Med. Chem. 2010, 18, 6080–6088. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, T.C.; Cunha-Oliveira, T.; Serafim, T.L.; Holy, J.; Krasutsky, D.; Kolomitsyna, O.; Krasutsky, P.; Moreno, A.M.; Oliveira, P.J. Dimethylaminopyridine derivatives of lupane triterpenoids cause mitochondrial disruption and induce the permeability transition. Bioorg. Med. Chem. 2013, 21, 7239–7249. [Google Scholar] [CrossRef] [PubMed]
- Shakurova, E.R.; Pozdnyakova, D.A.; Tretyakova, E.V.; Parfenova, L.V. One-Pot Synthesis of betulin triterpenoid quaternized pyridine derivatives and their antimicrobial activity. Lett. Drug. Des. Discov. 2020. [Google Scholar] [CrossRef]
- Parfenova, L.V.; Shakurova, E.R.; Ponedel’kina, I.Y. The method of synthesis of 1-(3-hydroxy,28-acetoxylup-20(29)-en-30-yl)–pyridinium bromide and its use as an antibacterial and antifungal agent. Patent application RU 2018 130 200 A, 20 August 2018. [Google Scholar]
- Shakurova, E.R.; Salimova, E.V.; Mescheryakova, E.S.; Parfenova, L.V. One-pot synthesis of quaternary pyridinium salts and tetrahydropyridine derivatives of fusidane triterpenoids. Chem. Heterocycl. Comp. 2019, 55. in press. [Google Scholar]
- Tang, Z.; Mayrargue, J.; Alami, M. Synthesis of piperidine derivatives by reduction of pyridinium salts. Synth. Commun. 2007, 37, 3367–3379. [Google Scholar] [CrossRef]
- Cockerill, F.R.; Wikler, M.A.; Alder, J.; Dudley, M.N.; Eliopoulos, G.M.; Ferraro, M.J.; Hardy, D.J.; Hecht, D.; Hindler, J.; Patel, J.; et al. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard, 9th ed.; Clinical and Laboratory Standards Institute: Wayne, NJ, USA, 2012. [Google Scholar]
- Zhadanov, R.I.; Golubev, V.A.; Rozantsev, E.G. Synthesis and structure of certain 1-oxopiperidinium tribromides. Izv. Akad. nauk SSSR. Ser. Khim. 1970, 1, 186–187. [Google Scholar] [CrossRef]
- Shibuya, M.; Tomizawa, M.; Iwabuchi, Y. Oxidative rearrangement of tertiary allylic alcohols employing oxoammonium salts. J. Org. Chem. 2008, 73, 4750–4752. [Google Scholar] [CrossRef] [PubMed]
- Percino, M.J.; Cerón, M.; Soriano-Moro, G.; Pacheco, J.A.; Castro, M.E.; Chapela, V.M.; Bonilla-Cruz, J.; Saldivar-Guerra, E. 2,2,6,6-Tetramethyl-1-oxopiperidinetribromide and two forms of 1-hydroxy-2,2,6,6-tetramethylpiperidinium bromide salt: syntheses, crystal structures and theoretical calculations. J. Mol. Struct. 2015, 1103, 254–264. [Google Scholar] [CrossRef]
- Urban, M.; Sarek, J.; Klinot, J.; Kornikova, G.; Hajduch, M. Synthesis of A-seco derivatives of betulinic acid with cytotoxic activity. J. Nat. Prod. 2004, 67, 1100–1105. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Comp. | Gram-Positive Bacteria | Gram-Negative Bacteria | Fungi | ||||
|---|---|---|---|---|---|---|---|
| Staphylococcus aureus | Escherichia coli | Klebsiella pneumoniae | Acinetobacter baumannii | Pseudomonas aeruginosa | Candida albicans | Cryptococcus neoformans | |
| 1 | 3.51 | −5.37 | 10.43 | 15.17 | 1.63 | 10.10 | −28.74 |
| 2 | 88.00 | 16.90 | 23.10 | 34.70 | 5.70 | 8.80 | −52.50 |
| 3 | 81.70 | 12.60 | −0.70 | −7.30 | −9.30 | 7.8 | −64.60 |
| Comp. | Gram-Positive Bacteria | Gram-Negative Bacteria | Fungi | ||||
|---|---|---|---|---|---|---|---|
| Staphylococcus aureus | Escherichia coli | Klebsiella pneumoniae | Acinetobacter baumannii | Pseudomonas aeruginosa | Candida albicans | Cryptococcus neoformans | |
| 2 | 4 | >32 | >32 | >32 | >32 | 32 | 32 |
| 3 | 16 | >32 | >32 | >32 | >32 | 32 | 32 |
| Fluconazole | 0.125 | 8 | |||||
| Colistin–sulfate | 0.125 | 0.25 | 0.25 | 0.25 | |||
| Vancomycin–HCl | 1 | ||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shakurova, E.R.; Parfenova, L.V. Synthesis of N-Heterocyclic Analogues of 28-O-Methyl Betulinate, and Their Antibacterial and Antifungal Properties. Molbank 2020, 2020, M1100. https://doi.org/10.3390/M1100
Shakurova ER, Parfenova LV. Synthesis of N-Heterocyclic Analogues of 28-O-Methyl Betulinate, and Their Antibacterial and Antifungal Properties. Molbank. 2020; 2020(1):M1100. https://doi.org/10.3390/M1100
Chicago/Turabian StyleShakurova, Elvira R., and Lyudmila V. Parfenova. 2020. "Synthesis of N-Heterocyclic Analogues of 28-O-Methyl Betulinate, and Their Antibacterial and Antifungal Properties" Molbank 2020, no. 1: M1100. https://doi.org/10.3390/M1100
APA StyleShakurova, E. R., & Parfenova, L. V. (2020). Synthesis of N-Heterocyclic Analogues of 28-O-Methyl Betulinate, and Their Antibacterial and Antifungal Properties. Molbank, 2020(1), M1100. https://doi.org/10.3390/M1100