Access to d- and l-Psicose Derivatives via Hydroxy Methylation of Ribono Lactone



Abstract
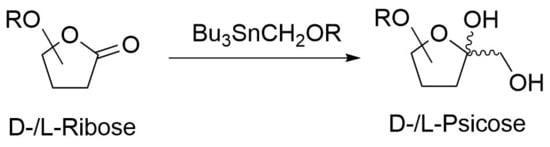
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Remarks
3.2. Synthesis of Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Wen, L.; Huang, K.; Wei, M.; Meisner, J.; Liu, Y.; Garner, K.; Zang, L.; Wang, X.; Li, X.; Fang, J.; et al. Facile Enzymatic Synthesis of Ketoses. Angew. Chem. Int. Ed. 2015, 54, 12654–12658. [Google Scholar] [CrossRef] [PubMed]
- Huwig, A.; Emmel, S.; Jäkel, G.; Giffhorn, F. Enzymatic synthesis of L-tagatose from galactitol with galactitol dehydrogenase from Rhodobacter sphaeroides D. Carbohydr. Res. 1997, 305, 337–339. [Google Scholar] [CrossRef]
- Li, Z.; Gao, Y.; Nakanishi, H.; Gao, X.; Cai, L. Biosynthesis of rare hexoses using microorganisms and related enzymes. Beilstein J. Org. Chem. 2013, 9, 2434–2445. [Google Scholar] [CrossRef] [PubMed]
- Ekeberg, D.; Morgenlie, S.; Stenstrøm, Y. Aldose–ketose interconversion in pyridine in the presence of aluminium oxide. Carbohydr. Res. 2007, 342, 1992–1997. [Google Scholar] [CrossRef]
- Doner, L.W. Isomerization of d-fructose by base: liquid-chromatographic evaluation and the isolation of d-psicose. Carbohydr. Res. 1979, 70, 209–216. [Google Scholar] [CrossRef]
- Mukaiyama, T.; Yuki, Y.; Suzuki, K. The steroselective synthesis of l-Tagatose-an application of Zn (II) mediated highly stereoselective addition of 2-furyllithium to polyoxygenated aldehyde. Chem. Lett. 1982, 11, 1169–1170. [Google Scholar]
- Matsumoto, T.; Enomoto, T.; Kurosaki, T. Facile synthesis of the next higher ketoses from aldoses. Chem. Commun. 1992, 610–611. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Raychaudhuri, U.; Chakraborty, R. Artificial sweeteners–a review. J. Food Sci. Technol. 2014, 51, 611–621. [Google Scholar] [CrossRef]
- Imrich, M.R.; Biehler, L.E.; Maichle-Mössmer, C.; Ziegler, T. Carbohydrate-Based Chiral Iodoarene Catalysts: A Survey through the Development of an Improved Catalyst Design. Molecules 2019, 24, 3883. [Google Scholar] [CrossRef]
- Imrich, M.R.; Kraft, J.; Maichle-Mössmer, C.; Ziegler, T. d-Fructose-based spiro-fused PHOX ligands: synthesis and application in enatioselective allylic alkylation. Beilstein J. Org. Chem. 2018, 14, 2082–2089. [Google Scholar] [CrossRef]
- Imrich, M.R.; Maichle-Mössmer, C.; Ziegler, T. d-Fructose based spiro-fused PHOX ligands: Palladium complexes and application in catalysis. Eur. J. Org. Chem. 2019, 3955–3963. [Google Scholar] [CrossRef]
- Imrich, M.R.; Ziegler, T. Carbohydrate based chiral iodoarene catalysts for enantioselective dearomative spirocyclization. Tetrahedron Lett. 2019, 60, 150954. [Google Scholar] [CrossRef]
- Price found at www.sigmaaldrich.com on 17 September 2019.
- Bols, M.; Szarek, W.A. Synthesis of 3-deoxy-3-fluoro-d-fructose. J. Chem. Soc. Chem. Commun. 1992, 445–446. [Google Scholar] [CrossRef]
- Bols, M.; Grubbe, H.; Jespersen, T.M.; Szarek, W.A. Hydroxymethylation of aldonolactones and a chemical synthesis of 3-deoxy-3-fluoro-d-fructose. Carbohydr. Res. 1994, 253, 195–206. [Google Scholar] [CrossRef]
- Shiozaki, M. Conversion of d-glucose to L-glucose: oxidative decarboxylation of alpha-oxycarboxylic acids via their diacyl peroxides. J. Org. Chem. 1991, 56, 528–532. [Google Scholar] [CrossRef]
- Martin, O.R.; Saavedra, O.M. Concise chemical synthesis of β-homonojirimycin and related compounds. Tetrahedron Lett. 1995, 36, 799–802. [Google Scholar] [CrossRef]
- Van Rijssel, E.R.; van Delft, P.; van Marle, D.V.; Bijvoets, S.M.; Lodder, G.; Overkleeft, H.S.; van der Marel, G.A.; Filippov, D.V.; Codée, J.D.C. Stereoselectivity in the Lewis Acid Mediated Reduction of Ketofuranoses. J. Org. Chem. 2015, 80, 4553–4565. [Google Scholar] [CrossRef]
- Meyer, N.; Seebach, D. Doppelt metalliertes Methanol. Alkohol-d1- und -d3-Reagenzien. Chem. Ber. 1980, 113, 1290–1303. [Google Scholar] [CrossRef]
- Corey, E.; Gras, J.L.; Ulrich, P. A new general method for protection of the hydroxyl function. Tetrahedron Lett. 1976, 17, 809–812. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Snyder, S.A.; Longbottom, D.A.; Nalbandian, A.Z.; Huang, X. New Uses for the Burgess Reagent in Chemical Synthesis: Methods for the Facile and Stereoselective Formation of Sulfamidates, Glycosylamines, and Sulfamides. Chem. Eur. J. 2004, 10, 5581–5606. [Google Scholar] [CrossRef]
- Lohse-Fraefel, N.; Carreira, E.M. Polyketide building blocks via diastereoselective nitrile oxide cycloadditions with homoallylic alcohols and monoprotected homoallylic diols. Chem. Eur. J. 2009, 15, 12065–12081. [Google Scholar] [CrossRef] [PubMed]
- Di Bussolo, V.; Fiasella, A.; Romano, M.R.; Favero, L.; Pineschi, M.; Crotti, P. Stereoselective synthesis of 2, 3-unsaturated-aza-O-glycosides via new diastereoisomeric N-Cbz-imino glycal-derived allyl epoxides. Org. Lett. 2007, 9, 4479–4482. [Google Scholar] [CrossRef] [PubMed]
- Price found at www.sigmaaldrich.com on 23 September 2019.





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imrich, M.R.; Ziegler, T. Access to d- and l-Psicose Derivatives via Hydroxy Methylation of Ribono Lactone. Molbank 2019, 2019, M1096. https://doi.org/10.3390/M1096
Imrich MR, Ziegler T. Access to d- and l-Psicose Derivatives via Hydroxy Methylation of Ribono Lactone. Molbank. 2019; 2019(4):M1096. https://doi.org/10.3390/M1096
Chicago/Turabian StyleImrich, Michael R., and Thomas Ziegler. 2019. "Access to d- and l-Psicose Derivatives via Hydroxy Methylation of Ribono Lactone" Molbank 2019, no. 4: M1096. https://doi.org/10.3390/M1096
APA StyleImrich, M. R., & Ziegler, T. (2019). Access to d- and l-Psicose Derivatives via Hydroxy Methylation of Ribono Lactone. Molbank, 2019(4), M1096. https://doi.org/10.3390/M1096