Abstract
2,3,5-Tri-O-benzyl- and 2,3,5-tri-O-methyl-d-ribono-γ-lactone were converted with (methoxyethoxymethoxy)methyl and benzyloxy tributylstannane into the corresponding protected d-psicoses as mixtures of anomers in 31%–72% yield. Treatment of 2,3,5-tri-O-methyl-l-ribono-γ-lactone with benzyloxy tributylstannane afforded the corresponding l-psicose derivative as an anomeric mixture in 72% yield. Both methylated psicoses were further converted into 1,2-O-isopropylidene-3,4,6-tri-O-methyl-d- and l-psicofuranosides, the respective α- and β-anomers of which could be separated and characterized.
1. Introduction
Only a few ketoses occur in nature in significant amounts. d-Fructose is the only ketose that occurs in large quantities in nature and can be isolated from plant material [1]. Other ketoses like psicose (Figure 1) are rare carbohydrates which cannot be isolated in considerable amounts from natural sources. They can be synthesized either by isomerization of the corresponding aldoses in low yields or by multi-step syntheses involving different protection and deprotection steps or by enzymatic methods [2,3,4,5,6,7,8] resulting in a high price for these ketoses. As part of an ongoing research project about the synthesis of carbohydrate derived catalysts for asymmetric syntheses [9,10,11,12] we required considerable amounts of d- and l-psicofuranose derivatives 3. Due to the high price of both enantiomers of psicose [13] we developed a specific synthetic route for the preparation of d- and l-psicose derivatives 3, starting from d- and l-ribose (2). We chose ribose as the starting material because both enantiomers of this sugar are commercially available at decent costs [13] and the stereocenters possess the same configuration as psicose.
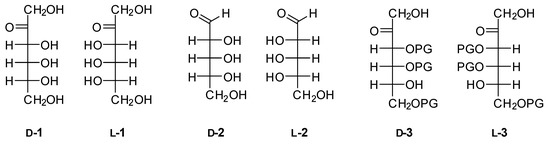
Figure 1.
Fischer projection of d- and l-psicose (1), d- and l-ribose (2) and d- and l-psicose derivatives 3. PG—protective group.
2. Results and Discussion
We first established a synthetic route starting with the cheap d-enantiomer of ribose and applied our findings later to the more expensive l-enantiomer. Since ribose is a pentose and psicose a hexose, the carbohydrate chain of the ribose had to be elongated by one carbon atom. Our strategy for accomplishing this was to first convert ribose to the corresponding lactone 6 and then elongate its carbon chain at C-1 via addition of the organometallic reagent 5 (Scheme 1). The latter could be obtained from tributylstannyl methanol 4 by transmetallation with butyl lithium and had already been used for the elongation of other carbohydrate derivatives [14,15,16,17].
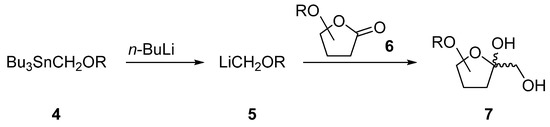
Scheme 1.
Planned elongation of the carbohydrate chain in order to derive hexose from a pentose.
At first we attempted to react known tri-O-benzyl ribono lactone (8) [18] with unprotected 4 (4a, R = H) [19] since this approach would have led directly to the target molecules without the need of any additional protecting group manipulations. Unfortunately, treatment of d-8 with 4a only resulted in decomposition of the starting materials. Next, we decided to use a suitably protected stannyl reagent 4 since such reagents had previously been used successfully for the elongation of other aldose derivatives. As a protecting group we chose the methoxyethoxmethyl group (MEM) due to its orthogonality to benzyl protecting groups and its facile removal [20]. In fact, treatment of lactone d-8 with MEM-protected 4b afforded the corresponding elongated sugar d-9, however in rather poor yield (Scheme 2).
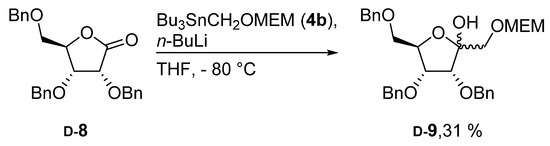
Scheme 2.
Hydroxy methylation of d-8 with methoxyethoxmethyl group (MEM)-protected organotin compound 4b.
It is known that stannyl reagents of the type 4 may rearrange upon transmetallation, with butyl lithium. Most likely this was the reason for the observed low yield. Such rearrangements may depend on the nature of the protecting group in 4. [19] Therefore, we decided to use a supposedly more stable benzyl group instead of the MEM protective group for reagent 4. In order to keep orthogonality between the protective groups at the ribono lactone and the stannyl reagent we also changed the substituents at the lactone accordingly. Here, we chose methyl groups as they can be introduced easily and are stable to broad range of reaction conditions. Thus known ribose derivative d-10 [21] was first oxidized to d-11 in excellent yield using iodine as the oxidant (Scheme 3). Addition of d-11 to a solution of the benzylated organotin reagent 4c and n-BuLi finally provided d-12 in good yield as an inseparable mixture of anomers.
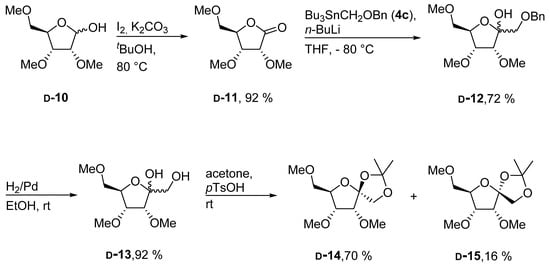
Scheme 3.
Synthesis of methylated d-psicose derivatives.
Next, the benzyl group at position 1 of psicose derivative d-12 was reductively removed in very high yield using palladium on charcoal as the catalyst. Deprotected compound d-13 again emerges as a mixture of inseparable anomers. In order to obtain an anomerically pure substance we further modified d-13 by converting it into the corresponding isopropylidene derivatives d-14 and d-15 both of which could be isolated in pure form. The configuration at the anomeric center of d-14 and d-15 was verified by NMR spectroscopy. The NOESY spectrum of d-14 shows a NOE between H-1 and the methyl groups at position 3 and 4 whereas the d-15 has a NOE between H-1 and H-3.
With a working synthetic sequence for the d-enantiomers in hand, we applied it to the l-enantiomer l-10 as well (Scheme 4). l-10 was prepared as described in the literature for the preparation of d-10. [21] As was expected, the synthetic route worked smoothly with yields nearly identical yields to those obtained for the d-enantiomers in all steps of the reaction sequence.
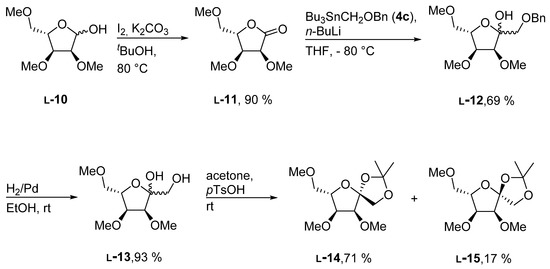
Scheme 4.
Synthesis of methylated l-psicose derivatives.
3. Materials and Methods
3.1. General Remarks
Reactions in dry solvents were carried out under an atmosphere of nitrogen. Dry THF was distilled from sodium/benzophenone, dry CH2Cl2 was distilled from P4O10, dry acetone was prepared from HPLC grade acetone by the addition of molecular sieves (4 Å). Tert-butanol and ethanol were HPLC grade and used without further purification. Solvents used for column chromatography were techniqual grade and distilled prior to their use. Petroleum ether (PE) refers to the fraction boiling at 60–90 °C. Silica gel “60 M” from Machery-Nagel was used for column chromatography. For reaction monitoring, TLC plates “Polygram Sil G/U254” from Machery-Nagel were used. Optical rotations were measured with a Perkin Elmer “Polarimeter 341”. NMR spectra were measured at a Bruker “Avance III HD 400” or a Bruker “Avance HD 300 NanoBay” and are calibrated to the solvent signal. For peak assignment additional spectra (DEPT, COSY, HMBC, HSQC) were recorded. The atoms are numbered in accordance with the carbohydrate nomenclature. High resolution mass spectra were measured at a Bruker “maXis 4G”. Elemental analysis was performed using a HEKAtech “Euro 3000 CHN”.
3.2. Synthesis of Compounds
[(Methoxy-ethoxy-methoxy)-methyl]tributylstannane (4b): To a solution of tributylstannyl-methanol (4a) (911 mg, 2.83 mmol) [22] in dry CH2Cl2 (10 mL) DIPEA (0.72 mL, 4.25 mmol) and MEM-Cl (0.49 mL, 4.25 mmol) were added and the reaction mixture was stirred at room temperature for 16 h. Then, again MEM-Cl (0.16 mL, 1.41 mmol) was added and the reaction mixture was stirred for additional 5 h. The reaction was quenched by the addition of MeOH and afterwards water was added. The aqueous phase was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried (Na2SO4) and the solvent was evaporated in vacuo. Column chromatography (PE + 2% Et3N → PE/EtOAc, 20/1 + 2% Et3N) provided 4b (985 mg, 85%) as colorless oil. Rf = 0.50 (PE/EtOAc, 10/1 + 2% Et3N); 1H-NMR (400 MHz, CDCl3) δ = 4.61 (s, 2H, SnCH2O), 3.76 (s, 2H, OCH2O), 3.62–3.68 (m, 2H, OCH2CH2O), 3.53–3.61 (m, 2H, OCH2CH2O), 3.33–3.45 (m, 3H, OCH3), 1.46–1.56 (m, 6H, C4H9), 1.26–1.35 (m, 6H, C4H9), 0.78–0.99 (m, 15H, C4H9); 13C-NMR (101 MHz, CDCl3) δ = 98.6 (SnCH2O), 71.8 (OCH2CH2O), 66.5 (OCH2CH2O), 59.0 (OCH3), 57.7 (OCH2O), 29.1 (CH2C3H7), 27.3 (CH2C3H7), 13.7 (CH3C3H6), 8.8 (CH2C3H7); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C17H38O3SnNa: 433.17351, found: 433.17360; Anal calcd for C17H38O3Sn: C 49.9, H 9.36, found: C 49.76, H 9.38.
3,4,6-Tri-O-benzyl-1-O-(methoxyethoxymethyl)-d-psicose (d-9): To a solution of 4b (239 mg, 0.58 mmol) in dry THF (2 mL) n-BuLi (348 μl, 0.56 mmol; 1.6 m in hexanes) and lactone d-8 (116 mg, 0.28 mmol; dissolved in 2 mL dry THF) were subsequently added at −80 °C. The reaction mixture was stirred at −80 °C for 40 min and afterwards the reaction was quenched by the addition of water (5 mL). The mixture was extracted with EtOAc (3 × 20 mL) and the combined organic layers were dried (Na2SO4). After evaporation of the solvent, the residue was purified by column chromatography (PE/EtOAc, 2/1 → 1/1), which provided d-9 (47 mg, 31%) as a colorless oil. = −15.6° (c = 1.0, CHCl3); Rf = 0.25 (PE/EtOAc, 2/1); 1H-NMR (400 MHz, CDCl3) δ = 7.11–7.36 (m, 15H, H-Ar), 5.60–5.77 (m, 1H, OH), 4.82–4.86 (m, 1H, CH2Ph), 4.66–4.69 (m, 1H, CH2Ph), 4.58–4.62 (m, 1H, H-1a), 4.54–4.58 (m, 1H, H-1b), 4.43–4.52 (m, 3H, CH2Ph), 4.35–4.42 (m, 1H, CH2Ph), 4.21–4.24 (m, 1H, H-5), 3.95–4.04 (m, 1H, H-6a), 3.64–3.75 (m, 2H, H-3, MEM), 3.44–3.63 (m, 4H, H-6b, MEM), 3.35–3.42 (m, 3H, H-4, MEM), 3.26 (s, 3H, CH3); 13C-NMR (101 MHz, CDCl3) δ = 137.8, 137.6, 137.5, 128.5, 128.4, 128.4, 128.1, 127.9, 127.9, 127.8, 127.4 (C-Ar), 97.5 (C-2), 95.9 (C-1), 75.4 (C-5), 75.1 (CH2Ph), 74.7 (C-3 or C-4), 73.3 (C-3 or C-4), 71.6 (MEM), 71.4, 71.2 (CH2Ph), 69.1, 66.9 (MEM), 58.9 (CH3), 57.5 (C-6); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C31H38O8Na: 561.24589, found: 561.24618; Anal calcd for C31H38O8: C 69.13, H 7.11, found: C 68.82, H 7.19.
2,3,5-Tri-O-methyl-d-ribono-1,4-lactone (d-11): A mixture of d-10 (3.83 g, 19.9 mmol), I2 (10.1 g, 39.8 mmol) and K2CO3 (5.51 g, 39.8 mmol) in tert-butanol (100 mL) was heated to 80 °C for 90 min. The reaction mixture was cooled to room temperature and water (10 mL) and solid Na2S2O4 were added to quench the excess of iodine. The mixture was filtered and the solvent was evaporated in vacuo. Column chromatography (PE/EtOAc, 2/1 → 1/1) provided d-11 (3.50 g, 92%) as light yellow oil. = +52.3° (c = 1.0, CHCl3); Rf = 0.59 (EtOAc); 1H-NMR (300 MHz, CDCl3) δ = 4.47–4.56 (m, 1H, H-4), 4.25 (d, J = 5.6 Hz, 1H, H-2), 4.05 (dd, J = 5.6, 1.7 Hz, 1H, H-3), 3.64 (s, 3H, CH3), 3.62 (t, J = 2.7 Hz, 2H, H-5), 3.48 (s, 3H, CH3), 3.31–3.39 (m, 3H, CH3); 13C-NMR (75 MHz, CDCl3) δ = 173.3 (C-1), 80.8 (C-4), 77.5 (C-3), 76.4 (C-2), 71.6 (C-5), 59.5, 59.2, 58.0 (CH3); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C8H14O5Na: 213.07334, found: 213.07374; Anal calcd for C8H14O5: C 50.52, H 7.42, found: C 50.12, H 7.32.
2,3,5-Tri-O-methyl-l-ribono-1,4-lactone (l-11): Prepared with the same protocol described for d-11, from l-10 (3.50 g, 18.2 mmol), I2 (9.24 g, 36.4 mmol) and K2CO3 (5.03 g, 36.4 mmol). Colorless oil, (3.12 g, 90%) yield after column chromatography. = −51.1° (c = 1.0, CHCl3); Rf and NMR data was identical to d-11; HRMS (ESI-TOF) m/z [M + Na]+: calcd for C8H14O5Na: 213.07334, found: 213.07350; Anal calcd for C8H14O5: C 50.52, H 7.42, found: C 50.88, H 7.71.
3,4,6-Tri-O-methyl-1-O-benzyl-d-psicose (d-12): To a solution of 4c (6.36 g, 15.5 mmol) [23] in dry THF (25 mL) n-BuLi (11.6 mL, 18.5 mmol; 1.6 m in hexanes) and lactone d-11 (1.96, 10.3 mmol; dissolved in 25 mL dry THF) was subsequently added at −80 °C. The reaction mixture was stirred at −80 °C for 90 min and afterwards allowed to reach room temperature and quenched by the addition of AcOH (1 mL), filtered and the solvent was evaporated in vacuo. Column chromatography (PE/EtOAc, 1/1) provided d-12 (2.32 g, 72%) as colorless oil. The product was isolated as mixture of anomers in ratio 4:1. = −6.7° (c = 1.0, CHCl3); Rf = 0.30 (PE/EtOAc, 1/1); 1H-NMR (300 MHz, CDCl3) δ = 7.23–7.41 (m, 5H, H-Ar, both anomers), 4.48–4.73 (m, 2H, CH2Ph, both anomers), 4.20–4.29 (m, 0.8H, H-5, major anomer), 4.07–4.17 (m, 0.2H, H-4, minor anomer), 4.01–4.05 (m, 0.2H, H-5, minor anomer), 3.89 (d, J = 5.1 Hz, 0.8H, H-3, major anomer), 3.78–3.85 (m, 0.8H, H-4, major anomer), 3.76 (d, J = 3.2 Hz, 0.2H, H-3, minor anomer), 3.29–3.68 (m, 13H, H-1a, H-1b, H-6a, H-6b, 3 × CH3); 13C-NMR (75 MHz, CDCl3) δ = 138.2, 128.3, 128.2, 127.9, 127.7, 127.6, 127.5 (C-Ar), 104.8 (C-2, minor anomer), 102.8 (C-2, major anomer), 82.9 (C-3, minor anomer), 80.7 (C-5, minor anomer), 79.9 (C-4, minor anomer), 79.8 (C-4, major anomer), 79.1 (C-3, major anomer), 79.0 (C-5, major anomer), 73.7 (CH2Ph, minor anomer), 73.5 (CH2Ph, major anomer), 73.0 (C-6, minor anomer), 72.5 (C-6, major anomer), 71.1 (C-1, major anomer), 70.8 (C-1, minor anomer), 60.0 (CH3, minor anomer), 59.3 (CH3, major anomer), 59.2 (CH3, minor anomer), 59.1 (CH3, major anomer), 58.4 (CH3, minor anomer), 58.4 (CH3, major anomer); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C16H24O6Na: 335.14651, found: 335.14699; Anal calcd for C16H24O6: C 61.52, H 7.74, found: C 61.05, H 7.84.
3,4,6-Tri-O-methyl-1-O-benzyl-l-psicose (l-12): Prepared with the same protocol as described for d-12, from 4c (8.76 g, 21.3 mmol), l-11 (2.70, 14.2 mmol) and n-BuLi (16.0 mL, 25.6 mmol; 1.6 m in hexanes). Colorless oil, (3.06 g, 69%) yield after column chromatography. = +7.3° (c = 1.0, CHCl3); Rf and NMR data was identical to d-11; HRMS (ESI-TOF) m/z [M + Na]+: calcd for C16H24O6Na: 335.14651, found: 335.14642; Anal calcd for C16H24O6: C 61.52, H 7.74, found: C 61.53, H 7.87.
3,4,6-Tri-O-methyl-d-psicose (d-13): A mixture of d-12 (2.64 g, 8.45 mmol) and Pd (200 mg, 0.19 mmol; 10% on activated carbon) in degassed EtOH (100 mL) was stirred under an atmosphere of hydrogen for 3 h. The reaction mixture was filtered through a pad of celite. After evaporation of the solvent in vacuo the residue was purified by column chromatography (EtOAc → EtOAc/i-PrOH 1/1) which provided d-13 (1.72 g, 92%) as a colorless oil. The product was isolated as mixture of anomers in ratio 4:1. = +1.6° (c = 1.0, CHCl3); Rf = 0.60 (EtOAc/i-PrOH, 1/1); 1H-NMR (400 MHz, CDCl3) δ = 4.32 (s, 0.8H, OH, major anomer), 4.19–4.22 (m, 0.8H, H-5, major anomer), 4.12–4.18 (m, 0.2H, H-5, minor anomer), 4.02–4.06 (m, 0.2H, H-4, minor anomer), 3.91–3.96 (m, 0.8H, H-4, major anomer), 3.87–3.90 (m, 0.8H, H-3, major anomer), 3.78–3.81 (m, 0.2H, H-3, minor anomer), 3.74 (s, 0.2H, OH, minor anomer), 3.32–3.68 (m, 13H, H-1a, H-1b, H-6a, H-6b, 3 × CH3), 2.50 (bs, 1H, OH); 13C-NMR (101 MHz, CDCl3) δ = 105.2 (C-2, minor anomer), 103.4 (C-2, major anomer), 83.9 (C-3, minor anomer), 80.7 (C-4, minor anomer), 80.2 (C-5, minor anomer), 80.2 (C-4, major anomer), 79.3 (C-3, major anomer), 79.2 (C-5, major anomer), 72.7 (C-6, minor anomer), 72.1 (C-6, major anomer), 64.8 (C-1, major anomer), 64.2 (C-1, minor anomer), 60.0 (CH3, minor anomer), 59.6 (CH3, major anomer), 59.3 (CH3, minor anomer), 59.3 (CH3, major anomer), 58.6 (CH3, major anomer), 58.5 (CH3, minor anomer); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C9H18O6Na: 245.09956, found: 245.09987; Anal calcd for C9H18O6: C 48.64, H 8.16, found: C 48.39, H 7.84.
3,4,6-Tri-O-methyl-l-psicose (l-13): Prepared with the same protocol as described for d-13, from l-12 (1.47 g, 4.70 mmol) and Pd (150 mg, 0.14 mmol; 10% on activated carbon). Colorless oil, (971 mg, 93%) yield after column chromatography. = −0.7° (c = 1.0, CHCl3); Rf and NMR data was identical to d-11; HRMS (ESI-TOF) m/z [M + Na]+: calcd for C9H18O6Na: 245.09956, found: 245.09987; Anal calcd for C9H18O6: C 48.64, H 8.16, found: C 48.83, H 8.24.
1,2-O-Isopropylidene-3,4,6-tri-O-methyl-α-d-psicofuranosid (d-14) and 1,2-O-isopropylidene-3,4,6-tri-O-methyl-β-d-psicofuranosid (d-15): To a solution of d-13 (26 mg, 0.117 mmol) in dry acetone (1 mL) para-toluene sulfonic acid (2.3 mg, 0.012 mmol; monohydrate) was added and the resulting mixture was stirred at room temperature for 20 h. The reaction mixture was alkalized by the addition of NH3 (0.1 mL; 25% in H2O) and the solvent was evaporated in vacuo. Column chromatography (PE/EtOAc, 1/1 → 1/3) afforded both anomers in two different fractions. Eluted first: d-14 (22 mg, 70%) as a colorless oil. = −35.3° (c = 1.0, CHCl3); Rf = 0.56 (PE/EtOAc, 1/1); 1H-NMR (400 MHz, CDCl3) δ = 4.26 (d, J = 9.7 Hz, 1H, H-1a), 4.05–4.09 (m, 1H, H-5), 3.97 (d, J = 9.7 Hz, 1H, H-1b), 3.78–3.83 (m, 1H, H-4), 3.76 (d, J = 4.5 Hz, 1H, H-3), 3.56 (s, 3H, CH3O), 3.50 (d, J = 5.1 Hz, 2H, H-6a, H-6b), 3.44 (s, 3H, CH3O), 3.40 (s, 3H, CH3O), 1.48 (s, 3H, CH3C), 1.40 (s, 3H CH3C); 13C-NMR (101 MHz, CDCl3) δ = 111.8 (C(CH3)2), 110.5 (C-2), 82.3 (C-3), 81.1 (C-4), 79.3 (C-5), 74.0 (C-6), 69.7 (C-1), 59.8, 59.4, 58.4 (CH3O), 26.3, 26.1 (C(CH3)2); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C12H22O6Na: 285.13086, found: 285.13089. Eluted second: d-15 (4.8 mg, 16%) as a colorless oil. = +10.5° (c = 1.0, CHCl3); Rf = 0.20 (PE/EtOAc, 1/1); 1H-NMR (400 MHz, CDCl3) δ = 4.23–4.26 (m, 1H, H-5), 4.08 (d, J = 9.1 Hz, 1H, H-1a), 4.03 (d, J = 9.1 Hz, 1H, H-1b), 3.77 (dd, J = 6.4, 2.9 Hz, 1H, H-4), 3.59 (d, J = 6.4 Hz, 1H, H-3), 3.52 (s, 3H, CH3O), 3.48 (d, J = 3.9 Hz, 2H, H-6a, H-6b), 3.44 (s, 3H, CH3O), 3.38 (s, 3H, CH3O), 1.53 (s, 3H, CH3C), 1.47 (s, 3H, CH3C); 13C-NMR (101 MHz, CDCl3) δ = 111.8 (C(CH3)2), 109.2 (C-2), 81.0 (C-5), 80.2 (C-3), 78.2 (C-4), 73.2 (C-6), 71.8 (C-1), 59.5, 58.7, 58.5 (CH3O), 26.6, 26.3 (C(CH3)2); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C12H22O6Na: 285.13086, found: 285.13106; Anal calcd for C12H22O6: C 54.95, H 8.45, found: C 54.47, H 8.28.
1,2-O-Isopropylidene-3,4,6-tri-O-methyl-α-l-psicofuranosid (l-14) and 1,2-O-isopropylidene-3,4,6-tri-O-methyl-β-l-psicofuranosid (l-15): Prepared with the same protocol as described for d-14 and d-15, from l-13 (148 mg, 0.666 mmol) and para-toluene sulfonic acid (12.6 mg, 0.066 mmol; monohydrate). Eluted first: l-14 (124 mg, 71%) as a colorless oil. = +37.6° (c = 1.0, CHCl3); Rf and NMR data was identical to d-14; HRMS (ESI-TOF) m/z [M + Na]+: calcd for C12H22O6Na: 285.13086, found: 285.13097; Anal calcd for C12H22O6: C 54.95, H 8.45, found: C 55.62, H 8:72. Eluted second: l-15 (30 mg, 17%) as a colorless oil. = −9.2° (c = 1.0, CHCl3); Rf and NMR data was identical to d-15; HRMS (ESI-TOF) m/z [M + Na]+: calcd for C12H22O6Na: 285.13086, found: 285.13110; Anal calcd for C12H22O6: 54.95, H 8.45, found: C 55.18, H 8.45.
4. Conclusions
In summary, we describe a method to prepare derivatives of both enantiomers of rare ketoses (i.e., psicose) from inexpensively ribose. The ketoses have the protecting groups necessary for the synthesis of ligands for asymmetric catalysis. With other inexpensive commercially available pentoses like for instance l-arabinose (100 g, 144 €) or d-xylose (1 kg, 57 €) derivatives of other rare ketoses like l-fructose (50 mg, 126 €), or d-sorbose (100 mg, 224 €) are accessible [24]. By using the benzylated lactone 8 in the sequence shown in Scheme 3 and Scheme 4, the completely deprotected psicose should be obtained after the reduction under an atmosphere of hydrogen. Since the methyl groups are hard to remove, other protective groups like MEM or TIPS at the carbohydrate may be used if an easily removable to benzyl orthogonal protective group is needed.
Supplementary Materials
Supplementary information (copies of NMR spectra) is available online.
Author Contributions
Experiment design, M.R.I. and T.Z.; experiment performance, M.R.I.; data interpretation, M.R.I.; writing—original draft preparation, M.R.I.; writing—review and editing, M.R.I., T.Z.; supervision, T.Z.
Funding
This research received no external funding.
Acknowledgments
The work of Anna Stroppel (proofreading the manuscript), Petra Schülzle (elemental analysis) and Dorothee Wistuba (HRMS) is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References and Notes
- Wen, L.; Huang, K.; Wei, M.; Meisner, J.; Liu, Y.; Garner, K.; Zang, L.; Wang, X.; Li, X.; Fang, J.; et al. Facile Enzymatic Synthesis of Ketoses. Angew. Chem. Int. Ed. 2015, 54, 12654–12658. [Google Scholar] [CrossRef] [PubMed]
- Huwig, A.; Emmel, S.; Jäkel, G.; Giffhorn, F. Enzymatic synthesis of L-tagatose from galactitol with galactitol dehydrogenase from Rhodobacter sphaeroides D. Carbohydr. Res. 1997, 305, 337–339. [Google Scholar] [CrossRef]
- Li, Z.; Gao, Y.; Nakanishi, H.; Gao, X.; Cai, L. Biosynthesis of rare hexoses using microorganisms and related enzymes. Beilstein J. Org. Chem. 2013, 9, 2434–2445. [Google Scholar] [CrossRef] [PubMed]
- Ekeberg, D.; Morgenlie, S.; Stenstrøm, Y. Aldose–ketose interconversion in pyridine in the presence of aluminium oxide. Carbohydr. Res. 2007, 342, 1992–1997. [Google Scholar] [CrossRef]
- Doner, L.W. Isomerization of d-fructose by base: liquid-chromatographic evaluation and the isolation of d-psicose. Carbohydr. Res. 1979, 70, 209–216. [Google Scholar] [CrossRef]
- Mukaiyama, T.; Yuki, Y.; Suzuki, K. The steroselective synthesis of l-Tagatose-an application of Zn (II) mediated highly stereoselective addition of 2-furyllithium to polyoxygenated aldehyde. Chem. Lett. 1982, 11, 1169–1170. [Google Scholar]
- Matsumoto, T.; Enomoto, T.; Kurosaki, T. Facile synthesis of the next higher ketoses from aldoses. Chem. Commun. 1992, 610–611. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Raychaudhuri, U.; Chakraborty, R. Artificial sweeteners–a review. J. Food Sci. Technol. 2014, 51, 611–621. [Google Scholar] [CrossRef]
- Imrich, M.R.; Biehler, L.E.; Maichle-Mössmer, C.; Ziegler, T. Carbohydrate-Based Chiral Iodoarene Catalysts: A Survey through the Development of an Improved Catalyst Design. Molecules 2019, 24, 3883. [Google Scholar] [CrossRef]
- Imrich, M.R.; Kraft, J.; Maichle-Mössmer, C.; Ziegler, T. d-Fructose-based spiro-fused PHOX ligands: synthesis and application in enatioselective allylic alkylation. Beilstein J. Org. Chem. 2018, 14, 2082–2089. [Google Scholar] [CrossRef]
- Imrich, M.R.; Maichle-Mössmer, C.; Ziegler, T. d-Fructose based spiro-fused PHOX ligands: Palladium complexes and application in catalysis. Eur. J. Org. Chem. 2019, 3955–3963. [Google Scholar] [CrossRef]
- Imrich, M.R.; Ziegler, T. Carbohydrate based chiral iodoarene catalysts for enantioselective dearomative spirocyclization. Tetrahedron Lett. 2019, 60, 150954. [Google Scholar] [CrossRef]
- Price found at www.sigmaaldrich.com on 17 September 2019.
- Bols, M.; Szarek, W.A. Synthesis of 3-deoxy-3-fluoro-d-fructose. J. Chem. Soc. Chem. Commun. 1992, 445–446. [Google Scholar] [CrossRef]
- Bols, M.; Grubbe, H.; Jespersen, T.M.; Szarek, W.A. Hydroxymethylation of aldonolactones and a chemical synthesis of 3-deoxy-3-fluoro-d-fructose. Carbohydr. Res. 1994, 253, 195–206. [Google Scholar] [CrossRef]
- Shiozaki, M. Conversion of d-glucose to L-glucose: oxidative decarboxylation of alpha-oxycarboxylic acids via their diacyl peroxides. J. Org. Chem. 1991, 56, 528–532. [Google Scholar] [CrossRef]
- Martin, O.R.; Saavedra, O.M. Concise chemical synthesis of β-homonojirimycin and related compounds. Tetrahedron Lett. 1995, 36, 799–802. [Google Scholar] [CrossRef]
- Van Rijssel, E.R.; van Delft, P.; van Marle, D.V.; Bijvoets, S.M.; Lodder, G.; Overkleeft, H.S.; van der Marel, G.A.; Filippov, D.V.; Codée, J.D.C. Stereoselectivity in the Lewis Acid Mediated Reduction of Ketofuranoses. J. Org. Chem. 2015, 80, 4553–4565. [Google Scholar] [CrossRef]
- Meyer, N.; Seebach, D. Doppelt metalliertes Methanol. Alkohol-d1- und -d3-Reagenzien. Chem. Ber. 1980, 113, 1290–1303. [Google Scholar] [CrossRef]
- Corey, E.; Gras, J.L.; Ulrich, P. A new general method for protection of the hydroxyl function. Tetrahedron Lett. 1976, 17, 809–812. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Snyder, S.A.; Longbottom, D.A.; Nalbandian, A.Z.; Huang, X. New Uses for the Burgess Reagent in Chemical Synthesis: Methods for the Facile and Stereoselective Formation of Sulfamidates, Glycosylamines, and Sulfamides. Chem. Eur. J. 2004, 10, 5581–5606. [Google Scholar] [CrossRef]
- Lohse-Fraefel, N.; Carreira, E.M. Polyketide building blocks via diastereoselective nitrile oxide cycloadditions with homoallylic alcohols and monoprotected homoallylic diols. Chem. Eur. J. 2009, 15, 12065–12081. [Google Scholar] [CrossRef] [PubMed]
- Di Bussolo, V.; Fiasella, A.; Romano, M.R.; Favero, L.; Pineschi, M.; Crotti, P. Stereoselective synthesis of 2, 3-unsaturated-aza-O-glycosides via new diastereoisomeric N-Cbz-imino glycal-derived allyl epoxides. Org. Lett. 2007, 9, 4479–4482. [Google Scholar] [CrossRef] [PubMed]
- Price found at www.sigmaaldrich.com on 23 September 2019.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).