1-Octyl-3-(3-(1-methylpyrrolidiniumyl)propyl)imidazolium Bis(trifluoromethane)sulfonimide




Abstract
:1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Information
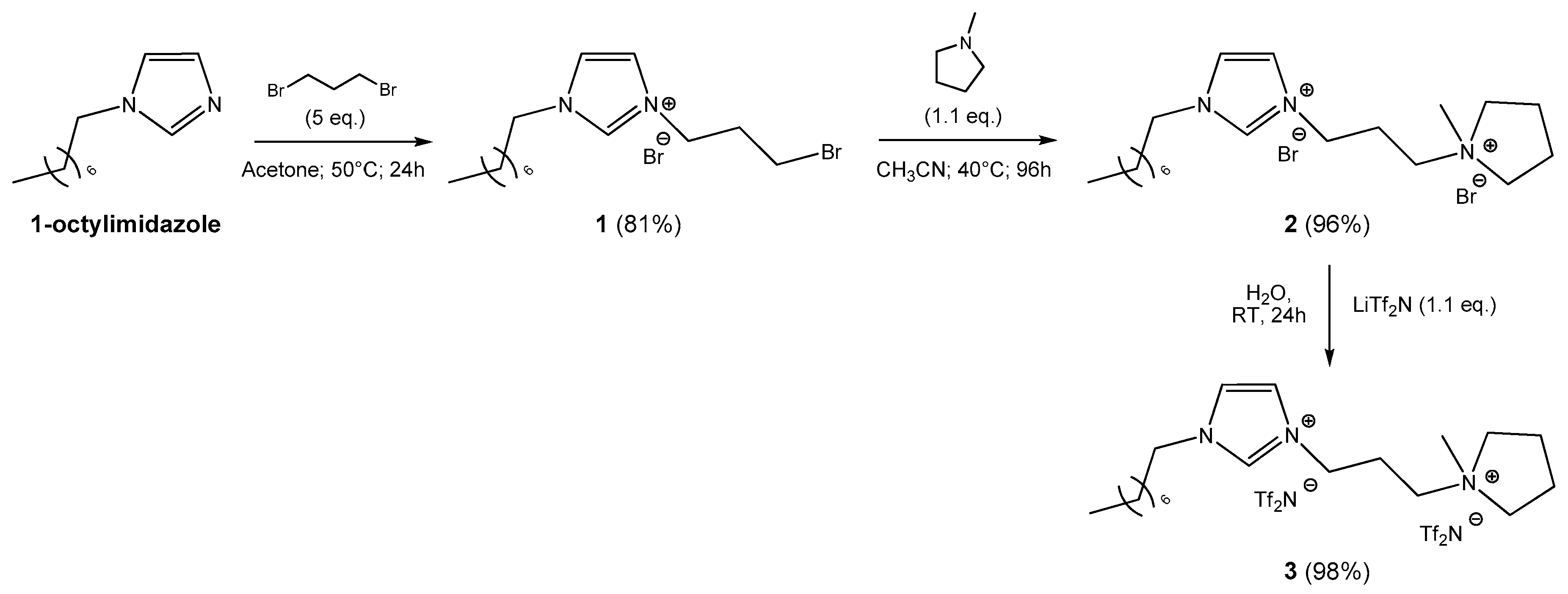
3.2. Synthesis of Ionic Liquid 1
3.3. Synthesis of Ionic Liquid 2
3.4. Synthesis of Ionic Liquid 3
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Welton, T. Ionic liquid: A brief history. Biophys. Rev. 2018, 10, 691–706. [Google Scholar] [CrossRef]
- Tokuda, H.; Hayamizu, K.; Ishii, K.; Susan, M.A.B.H.; Watanabe, M. Physicochemical Properties and Structures of Room Temperature Ionic Liquids. 2. Variation of Alkyl Chain Length in Imidazolium Cation. J. Phys. Chem. B 2005, 109, 6103–6110. [Google Scholar] [CrossRef]
- Chen, Q.; Wu, K.; He, C. Thermal Conductivity of Ionic Liquids at Atmospheric Pressure: Database, Analysis, and Prediction Using a Topological Index Method. Ind. Eng. Chem. Res. 2014, 53, 7224–7232. [Google Scholar] [CrossRef]
- Zech, O.; Stoppa, A.; Buchner, R.; Kunz, W. The Conductivity of Imidazolium-Based Ionic Liquids from (248 to 468) K. B. Variation of the Anion. J. Chem. Eng. Data 2010, 55, 1774–1778. [Google Scholar] [CrossRef]
- Chiappe, C.; Margari, P.; Mezzetta, A.; Pomelli, C.S.; Koutsoumpos, S.; Papamichael, M.; Giannios, P.; Moutzouris, K. Temperature effects on the viscosity and the wavelength-dependent refractive index of imidazolium-based ionic liquids with a phosphorus-containing anion. Phys. Chem. Chem. Phys. 2017, 19, 8201–8209. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.; Qin, L.; Jiang, J.; Mu, T.; Gao, G. Thermal, electrochemical and radiolytic stabilities of ionic liquids. Phys. Chem. Chem. Phys. 2018, 20, 8382–8402. [Google Scholar] [CrossRef] [PubMed]
- Hallett, J.P.; Welton, T. Room-Temperature Ionic Liquids: Solvents for Synthesis and Catalysis. 2. Chem. Rev. 2011, 5, 3508–3576. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, S.; Deng, Y. Recent advances in ionic liquid catalysis. Green Chem. 2011, 13, 2619–2637. [Google Scholar] [CrossRef]
- Radai, Z.; Kiss, N.Z.; Keglevich, G. An Overview of the Applications of Ionic Liquids as Catalysts and Additives in Organic Chemical Reactions. Curr. Org. Chem. 2018, 22, 533–556. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, Z.; Sun, X.-G.; Hu, Y.-S.; Xing, H.; Dai, S. Ionic liquids and derived materials for lithium and sodium batteries. Chem. Soc. Rev. 2018, 47, 2020–2064. [Google Scholar] [CrossRef]
- Longhi, M.; Arnaboldi, S.; Husanu, E.; Grecchi, S.; Buzzi, I.F.; Cirilli, R.; Rizzo, S.; Chiappe, C.; Mussini, P.R.; Guazzelli, L. A family of chiral ionic liquids from the natural pool: Relationships between structure and functional properties and electrochemical enantiodiscrimination tests. Electrochim. Acta 2019, 298, 194–209. [Google Scholar] [CrossRef]
- Isik, M.; Sardon, H.; Mecerreyes, D. Ionic Liquids and Cellulose: Dissolution, Chemical Modification and Preparation of New Cellulosic Materials. Int. J. Mol. Sci. 2014, 15, 11922–11940. [Google Scholar] [CrossRef] [PubMed]
- Mezzetta, A.; Becherini, S.; Pretti, C.; Monni, G.; Casu, V.; Chiappe, C.; Guazzelli, L. Insights into the levulinate-based ionic liquid class: Synthesis, cellulose dissolution evaluation and ecotoxicity assessment. New J. Chem. 2019, 43, 13010–13019. [Google Scholar] [CrossRef]
- Palazzo, I.; Mezzetta, A.; Guazzelli, L.; Sartini, S.; Pomelli, C.S.; Parker, W.O., Jr.; Chiappe, C. Chiral ionic liquids supported on natural sporopollenin microcapsules. RSC Adv. 2018, 8, 21174–21183. [Google Scholar] [CrossRef]
- Mezzetta, A.; Łuczak, J.; Woch, J.; Chiappe, C.; Nowicki, J.; Guazzelli, L. Surface active fatty acid ILs: Influence of the hydrophobic tail and/or the imidazolium hydroxyl functionalization on aggregates formation. J. Mol. Liq. 2019, 289, 111155. [Google Scholar] [CrossRef]
- Nowicki, J.; Woch, J.; Łuczak, J.; Zarębsk, M.; Nowakowska-Bogdan, E.; Mościpan, M. Micellar Route of the Synthesis of Alkyl Xylosides: An Unexpected Effect of Amphiphilic Imidazolium Ionic Liquids. ChemistrySelect 2018, 3, 5254–5262. [Google Scholar] [CrossRef]
- Yoshizawa, M.; Ito-Akita, K.; Ohno, H. Evidence of interaction between anion and polyether in the bulk. Electrochim. Acta 2000, 45, 1617–1621. [Google Scholar] [CrossRef]
- Anderson, J.L.; Ding, R.; Ellern, A.; Armstrong, D.W. Structure and Properties of High Stability Geminal Dicationic Ionic Liquids. J. Am. Chem. Soc. 2005, 127, 593–604. [Google Scholar] [CrossRef]
- Guglielmero, L.; Mezzetta, A.; Guazzelli, L.; Pomelli, C.S.; D’Andrea, F.; Chiappe, C. Systematic synthesis and properties evaluation of dicationic ionic liquids, and a glance into a potential new field. Front. Chem. 2018, 6, 612. [Google Scholar] [CrossRef]
- Shirota, H.; Mandai, T.; Fukazawa, H.; Kato, T. Comparison between Dicationic and Monocationic Ionic Liquids: Liquid Density, Thermal Properties, Surface Tension, and Shear Viscosity. J. Chem. Eng. Data 2011, 56, 2453–2459. [Google Scholar] [CrossRef]
- Pitawala, J.; Matic, A.; Martinelli, A.; Jacobsson, P.; Koch, V.; Croce, F. Thermal Properties and Ionic Conductivity of Imidazolium Bis(trifluoromethanesulfonyl)imide Dicationic Ionic Liquids. J. Phys. Chem. B 2009, 113, 10607–10610. [Google Scholar] [CrossRef] [PubMed]
- Talebi, M.; Patil, R.A.; Sidisky, L.M.; Berthod, A.; Armstrong, D.W. Branched-chain dicationic ionic liquids for fatty acid methyl ester assessment by gas chromatography. Anal. Bioanal. Chem. 2018, 410, 4633–4643. [Google Scholar] [CrossRef] [PubMed]
- Guglielmero, L.; Mezzetta, A.; Pomelli, C.S.; Chiappe, C.; Guazzelli, L. Evaluation of the effect of the dicationic ionic liquid structure on the cycloaddition of CO2 to epoxides. J. CO2 Util. 2019, 34, 437–445. [Google Scholar] [CrossRef]
- Cognigni, A.; Kampichler, S.; Bica, K. Surface-active ionic liquids in catalysis: Impact of structure and concentration on the aerobic oxidation of octanol in water. J. Colloid Interface Sci. 2017, 492, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, C.; D’Anna, F.; Marullo, S.; Noto, R. Task Specific Dicationic Ionic Liquids: Recyclable Reaction Media for the Mononuclear Rearrangement of Heterocycles. J. Org. Chem. 2014, 79, 8678–8683. [Google Scholar] [CrossRef]
- Rizzo, C.; Mandoli, A.; Marullo, S.; D’Anna, F. Ionic Liquid Gels: Supramolecular Reaction Media for the Alcoholysis of Anhydrides. J. Org. Chem. 2019, 10, 6356–6365. [Google Scholar] [CrossRef]
- Mei, X.; Yue, Z.; Ma, Q.; Dunya, H.; Mandal, B.K. Synthesis and electrochemical properties of new dicationic ionic liquids. J. Mol. Liq. 2018, 272, 1001–1018. [Google Scholar] [CrossRef]
- Salimi, P.; Kowsari, E. Electrochemical Study of Li-Ion 18650 Cylindrical Rechargeable Cell at Elevated Temperature Using Geminal Dicationic Ionic Liquid as Electrolyte Additive. J. Electron. Mater. 2019, 48, 2254–2262. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, T.H.; Kim, D.Y.; Park, N.G.; Ahn, K.D. Novel thixotropic gel electrolytes based on dicationic bis-imidazolium salts for quasi-solid-state dye-sensitized solar cells. J. Power Sources 2008, 175, 692–697. [Google Scholar] [CrossRef]
- Xiaohui, Z.; Dong, A.; Zhiwen, Y. Adsorption and thermodynamic properties of dissymmetric gemini imidazolium surfactants with different spacer length. J. Disper. Sci. Technol. 2016, 38, 296–302. [Google Scholar] [CrossRef]
- Mezzetta, A.; Perillo, V.; Guazzelli, L.; Chiappe, C. Thermal Behavior analysis a valuable tool for comparing ionic liquids of different classes. J. Therm. Anal. Calorim. 2019. [Google Scholar] [CrossRef]
- Cao, Y.; Mu, T. Comprehensive investigation on the thermal stability of 66 ionic liquids by thermogravimetric analysis. Ind. End. Chem. Res. 2014, 53, 8651–8664. [Google Scholar] [CrossRef]

{kind=link}
| ILs | Water | Methanol | Hexane | Acetonitrile | Dichloromethane | Density (g/cm3) |
|---|---|---|---|---|---|---|
| C3Br(C8Im)/Br | X | X | - | X | X | 1.07 |
| C3(C8Im)(C1Pyrr)/2 Br | X | X | - | X | - | 1.10 |
| C3(C8Im)(C1Pyrr)/2 Tf2N | - | X | - | X | X | 1.47 |
| IL | Shear Rate (1/s) | Viscosity (mPa s) |
|---|---|---|
| C3Br(C8Im)/Br | 1.12 | 11197 |
| 12.5 | 9599.8 | |
| 99.9 | 3597.9 | |
| C3(C8Im)(C1Pyrr)/2 Tf2N | 1.12 | 1133.7 |
| 12.5 | 932.47 | |
| 99.9 | 811.14 |
| Compound | 1 | 2 | 3 | |
|---|---|---|---|---|
| IL | C3Br(C8Im)/Br | C3(C8Im)(C1Pyrr)/2 Br | C3(C8Im)(C1Pyrr)/2 Tf2N | |
| TGA | Tstart 5% (°C) | 263.1 | 291.4 | 393.4 |
| Tonset (°C) | 282.1 | 304.8 | 411.3 | |
| Tpeak (°C) | 333.3 | 339.7 | 433.2 | |
| DSC | Tg (°C) | −50.3 | - | −51.5 |
| Tm (°C) | - | 113.7 | - | |
| ΔH (KJ/mol) | - | 13.61 | - | |
| Ts-s (°C) | - | 50.2(c)/50.3 (h) | - | |
| ΔH (KJ/mol) | - | −4.378 (c)/4.911 (h) | - | |
| Tc (°C) | - | 101.2 | - | |
| ΔH (KJ/mol) | - | −14.57 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mezzetta, A.; Pomelli, C.S.; D’Andrea, F.; Guazzelli, L. 1-Octyl-3-(3-(1-methylpyrrolidiniumyl)propyl)imidazolium Bis(trifluoromethane)sulfonimide. Molbank 2019, 2019, M1089. https://doi.org/10.3390/M1089
Mezzetta A, Pomelli CS, D’Andrea F, Guazzelli L. 1-Octyl-3-(3-(1-methylpyrrolidiniumyl)propyl)imidazolium Bis(trifluoromethane)sulfonimide. Molbank. 2019; 2019(4):M1089. https://doi.org/10.3390/M1089
Chicago/Turabian StyleMezzetta, Andrea, Christian S. Pomelli, Felicia D’Andrea, and Lorenzo Guazzelli. 2019. "1-Octyl-3-(3-(1-methylpyrrolidiniumyl)propyl)imidazolium Bis(trifluoromethane)sulfonimide" Molbank 2019, no. 4: M1089. https://doi.org/10.3390/M1089
APA StyleMezzetta, A., Pomelli, C. S., D’Andrea, F., & Guazzelli, L. (2019). 1-Octyl-3-(3-(1-methylpyrrolidiniumyl)propyl)imidazolium Bis(trifluoromethane)sulfonimide. Molbank, 2019(4), M1089. https://doi.org/10.3390/M1089