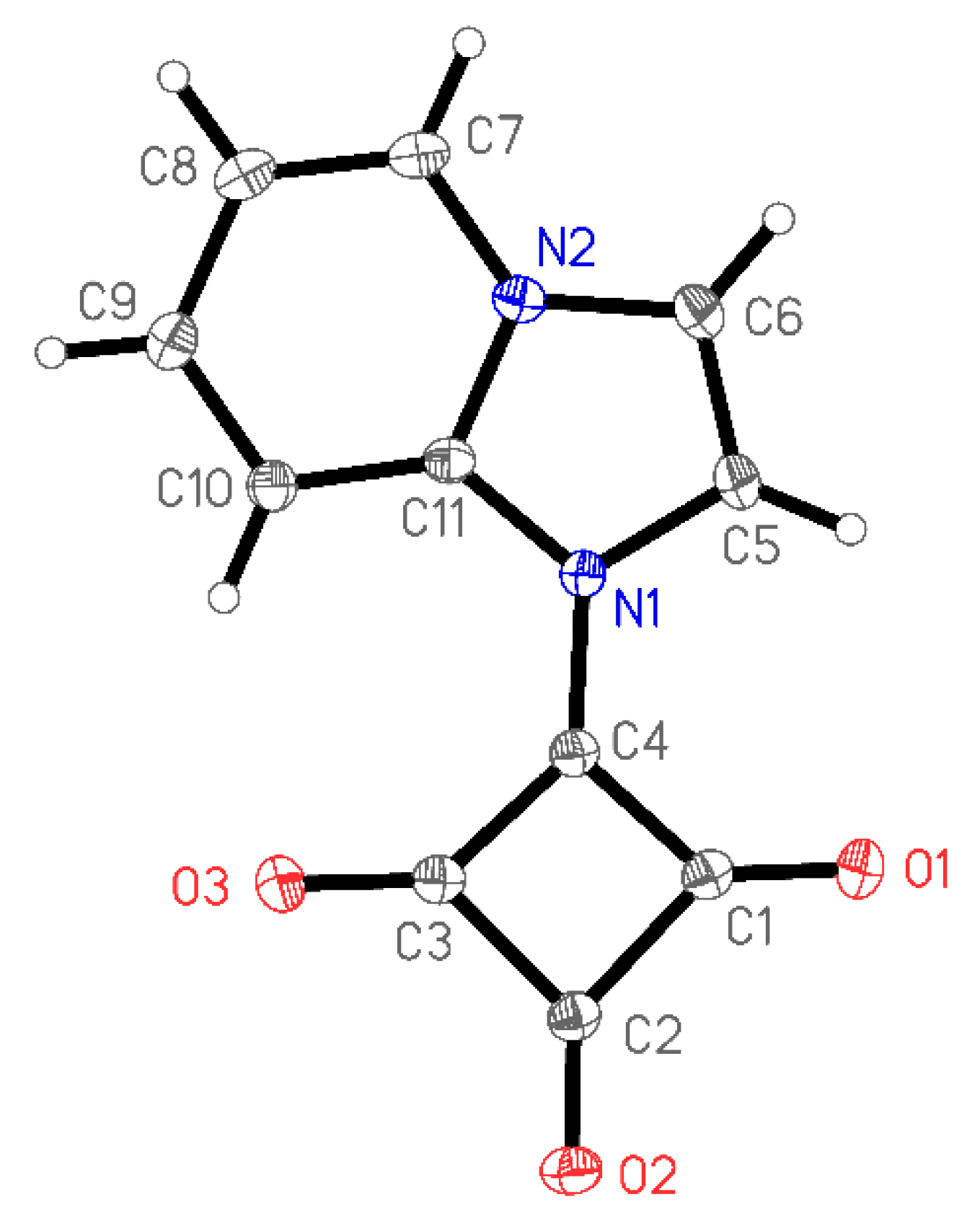
1-(Imidazo[1,2-a]pyridin-1-ium-1-yl)-2,3,4-trioxocyclobutan-1-ide


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
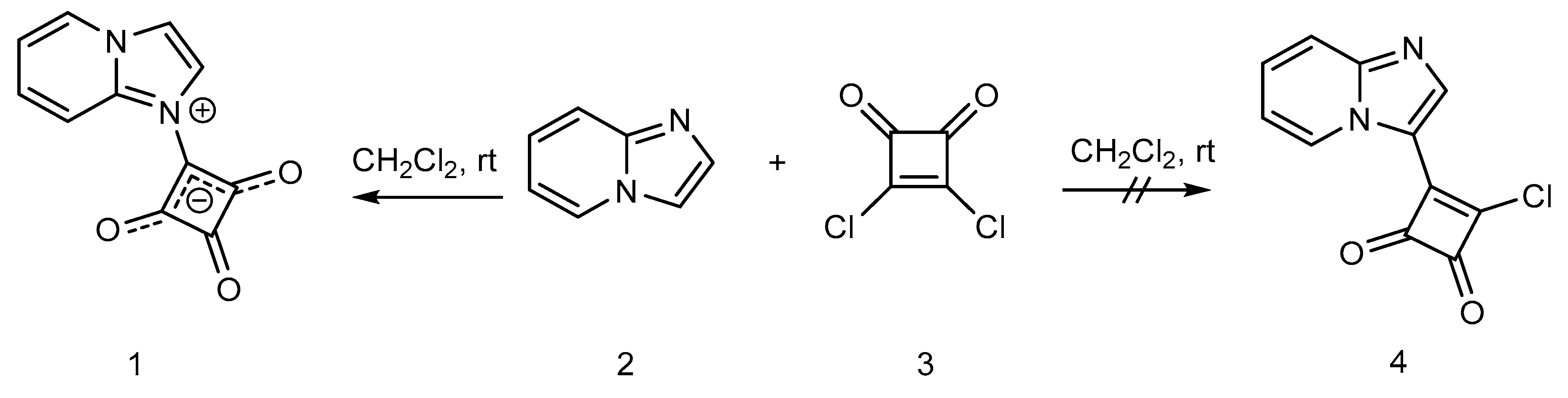
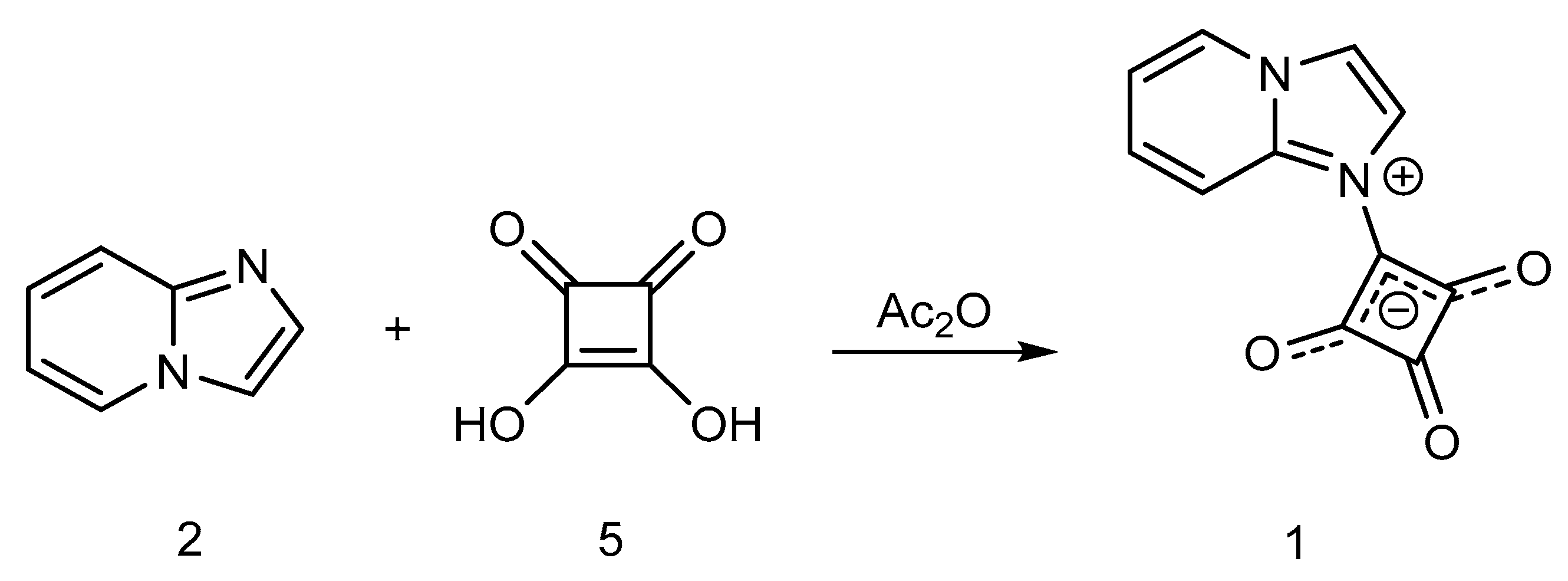
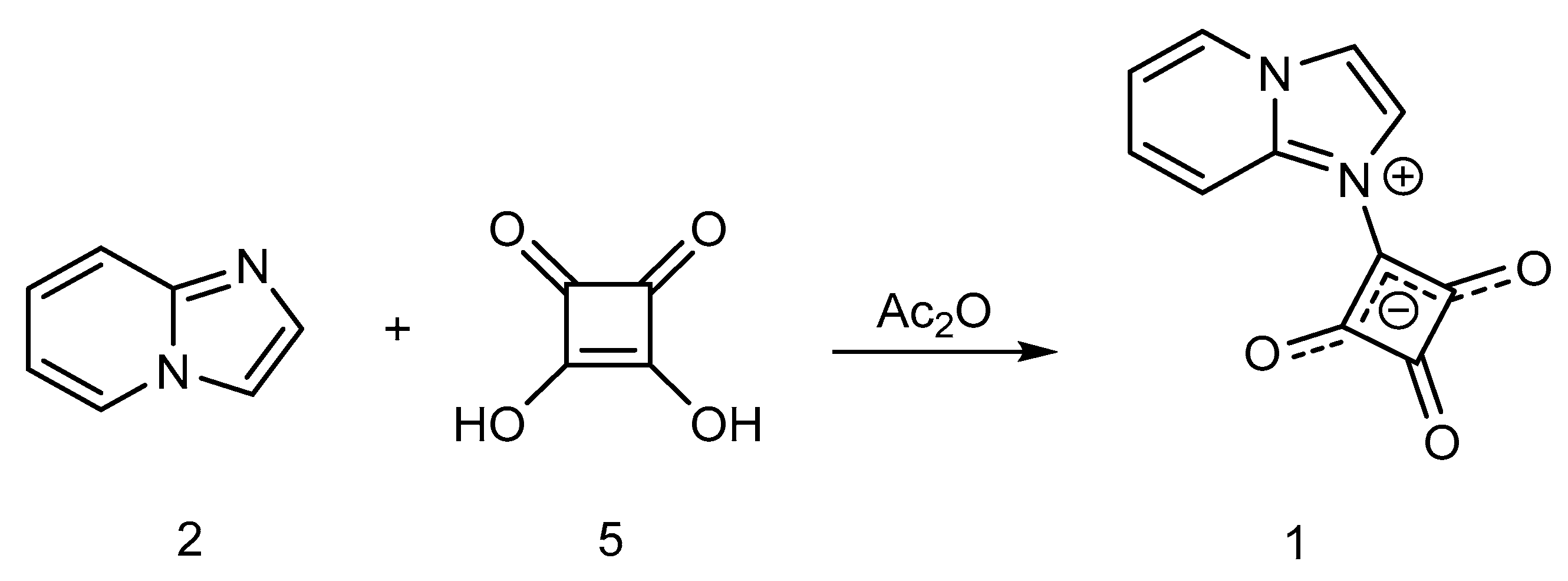
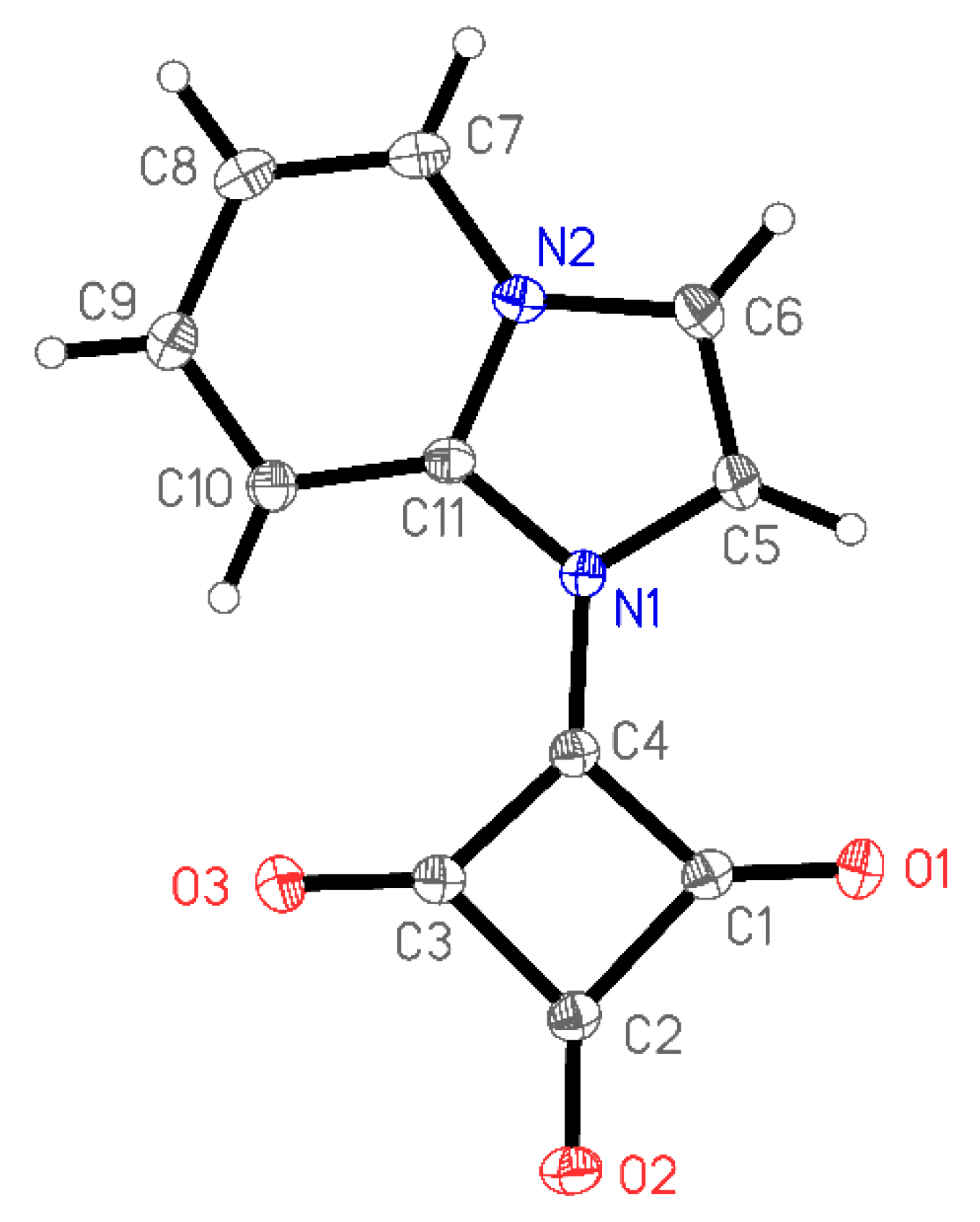
2. Results and Discussion
3. Materials and Methods
3.1. Instrumentation
3.2. Syntheses
Synthesis of 1-(Imidazo[1,2-a]pyridin-1-ium-1-yl)-2,3,4-trioxocyclobutan-1-ide (1); Method 1
Synthesis of 1-(Imidazo[1,2-a]pyridin-1-ium-1-yl)-2,3,4-trioxocyclobutan-1-ide (1); Method 2
3.3. X-Ray Structure Determination of 1
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kunick, C.; Lande, H.; Gruenefeld, J.; Dzikowski, R.; Nasereddin, A. New Indole Compounds Having Antiprotozoal Activity and Its Use as Well as Methods for Producing the Same. WO/2017/008826. 2017. [Google Scholar]
- Schmidt, A.H.; Thiel, S.H.; Gaschler, O. Oxocarbons and related compounds. Part 24. Chlorosquarylation of indoles. J. Chem. Soc. Perkin Trans. 1 1996, 495–496. [Google Scholar] [CrossRef]
- Lande, D.H.; Messal, M.; Reimann, V.; Renger, C.; Rurka, C.; Schoemaker, J.; Schröder, N.K.; Grünefeld, J. 3-Chloro-4-(5-methoxy-1H-indol-3-yl)cyclobut-3-ene-1,2-dione. Molbank 2013, 2013, M805. [Google Scholar] [CrossRef]
- Lande, H.; Kunick, C.; Grünefeld, J. 2,3,4-Trioxo-1-(1H-pyrrolo[2,3-b]pyridin-7-ium-7-yl)-cyclobutan-1-ide. Molbank 2018, 2018, M1026. [Google Scholar] [CrossRef]
- Chayer, S.; Schmitt, M.; Collot, V.; Bourguignon, J.-J. Regiospecific thermal C-acylation of imidazo[1,2a]pyridines via an N-acylimidazolium intermediate. Tetrahedron Lett. 1998, 39, 9685–9688. [Google Scholar] [CrossRef]
- Schmidt, A.H.; Schneider, M.; Aimène, A.; Straus, M.; Botzet, D. Oxokohlenstoffe und verwandte Verbindungen, IX. Zur Dreikomponentenreaktion Quadratsäuredichlorid–Amin–Wasser: Weitere Stickstoff-Betaine der Quadratsäure sowie Bildung von Ammoniumsalzen des Quadratsäuremonochlorides und von Quadratsäureamidchloriden. Chem.-Ztg. 1985, 109, 333–339. [Google Scholar]
- Schmidt, A.H.; Becker, U.; Aimène, A. Ein neues, einfaches Verfahren zur Darstellung von Ammonium- und Phosphonium-Betainen der Quadratsäure. Tetrahedron Lett. 1984, 25, 4475–4478. [Google Scholar] [CrossRef]
- Korkmaz, U.; Bulut, I.; Bulut, A. Crystal structure of 2-ethyl-4-methyl-1-(2-oxido-3,4-dioxocyclobut-1-en-1-yl)-1H-imidazol-3-ium. Acta Crystallogr. 2016, E72, 998–1001, (Refcode AQAGOC). [Google Scholar] [CrossRef] [PubMed]
- Petrov, P.; Deligeorgiev, T.; Ivanova, B. CCDC-1585680. CSD Commun. 2017. (Refcode XEHYUH). [Google Scholar] [CrossRef]
- Kolev, T.; Wortmann, R.; Spitelier, M.; Sheldrick, W.S.; Mayer-Figge, H. 4-Phenylpyridinium betaine of squaric acid. Acta Crystallogr. 2005, E61, o1090–o1092, (Refcode YARYUM). [Google Scholar] [CrossRef]
- Song, J.-L.; Yi, F.-Y.; Mao, J.-G. New types of luminescent lanthanide squarato-aminophosphonates. Cryst. Growth Des. 2009, 9, 3273–3277. [Google Scholar] [CrossRef]
- Diffractometer Software. CrysalisPro; Version 1.131.38.43c; Rigaku Oxford Diffraction: Yarnton, UK, 2015. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grünefeld, J.; Kunick, C.; Jones, P.G. 1-(Imidazo[1,2-a]pyridin-1-ium-1-yl)-2,3,4-trioxocyclobutan-1-ide. Molbank 2019, 2019, M1072. https://doi.org/10.3390/M1072
Grünefeld J, Kunick C, Jones PG. 1-(Imidazo[1,2-a]pyridin-1-ium-1-yl)-2,3,4-trioxocyclobutan-1-ide. Molbank. 2019; 2019(3):M1072. https://doi.org/10.3390/M1072
Chicago/Turabian StyleGrünefeld, Johann, Conrad Kunick, and Peter G. Jones. 2019. "1-(Imidazo[1,2-a]pyridin-1-ium-1-yl)-2,3,4-trioxocyclobutan-1-ide" Molbank 2019, no. 3: M1072. https://doi.org/10.3390/M1072
APA StyleGrünefeld, J., Kunick, C., & Jones, P. G. (2019). 1-(Imidazo[1,2-a]pyridin-1-ium-1-yl)-2,3,4-trioxocyclobutan-1-ide. Molbank, 2019(3), M1072. https://doi.org/10.3390/M1072