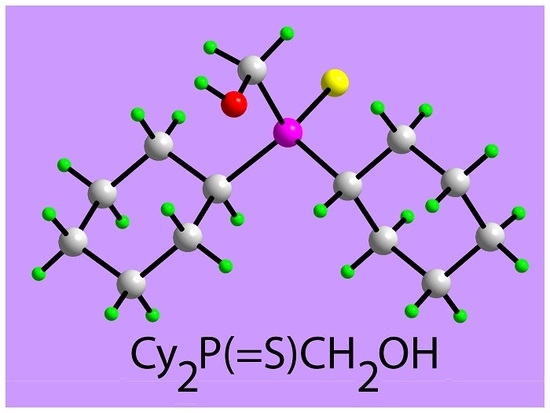
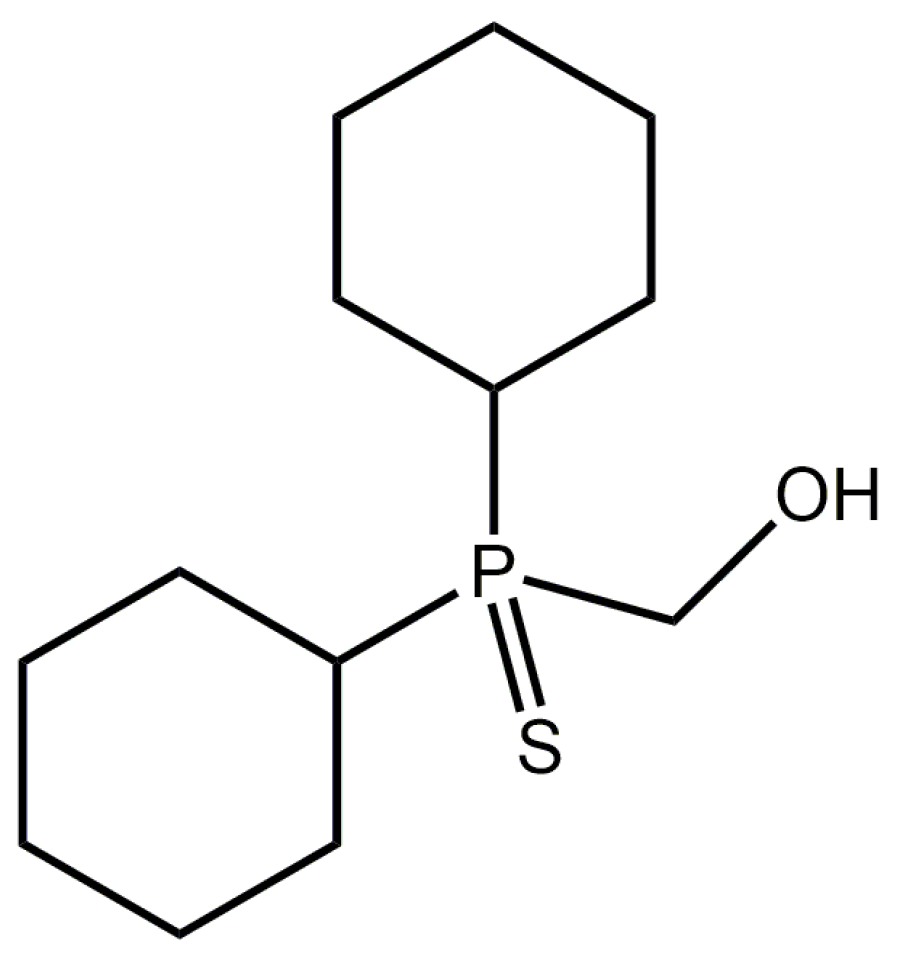
[Dicyclohexyl(sulfanylidene)-λ5-phosphanyl]methanol

 , ,
, , 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
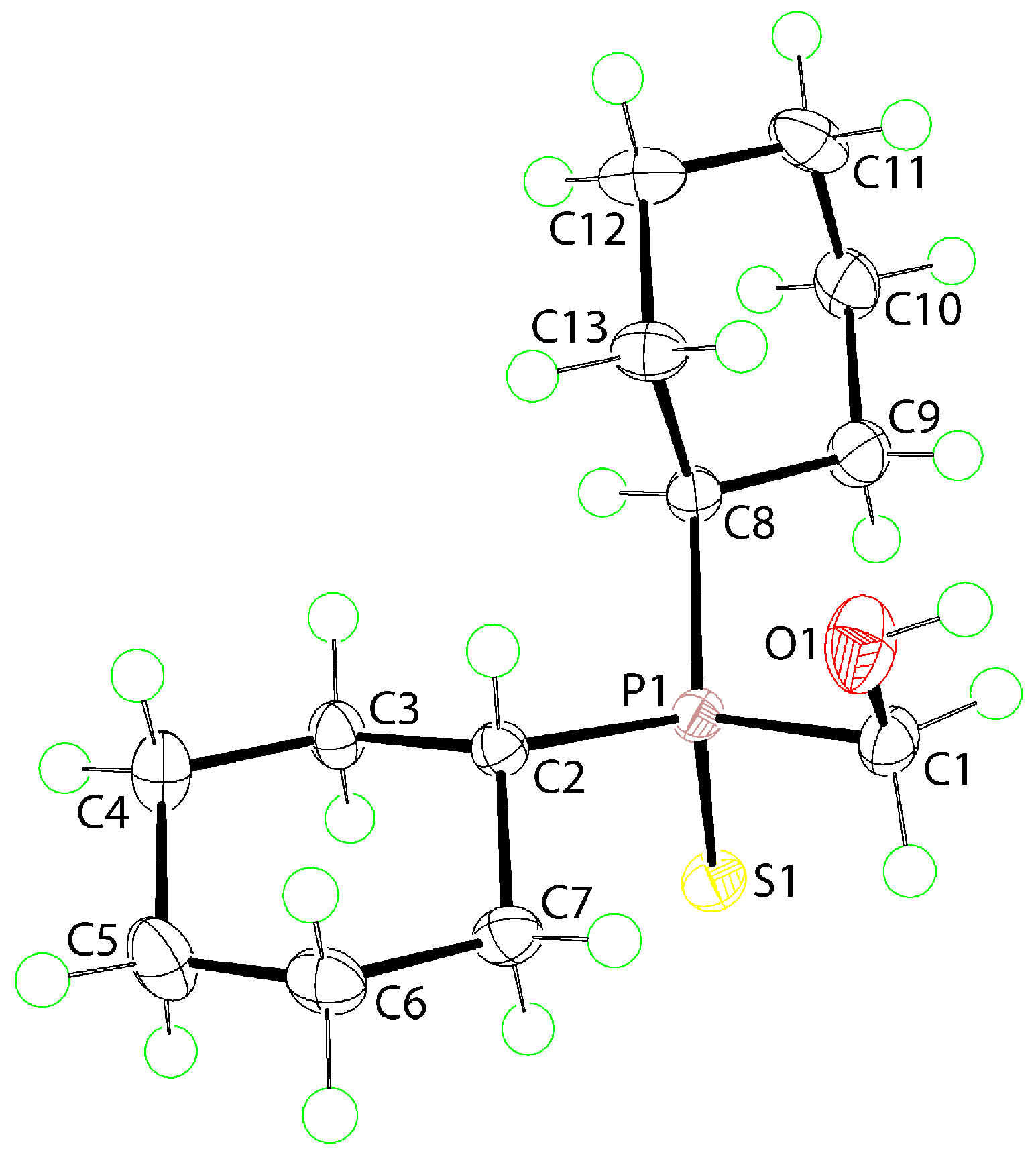
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthesis Cy2P(S)CH2OH (1)
3.3. Crystallography
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- James, B.R.; Lorenzini, F. Developments in the chemistry of tris(hydroxymethyl)phosphine. Coord. Chem. Rev. 2010, 254, 420–430. [Google Scholar] [CrossRef]
- Henderson, W.; Alley, S.R. Ferrocenyl hydroxymethylphosphines (η5-C5H5)Fe[η5-C5H4P(CH2OH)2] and 1,1′-[Fe{η5-C5H4P(CH2OH)2}2] and their chalcogenide derivatives. J. Organomet. Chem. 2002, 658, 181–190. [Google Scholar] [CrossRef]
- Goodwin, N.J.; Henderson, W.; Nicholson, B.K.; Sarfo, J.K.; Fawcett, J.; Russell, D.R. Synthesis and reactivity of the ferrocene-derived phosphine [Fe(η-C5H5){η-C5H4CH2P(CH2OH)2}]. J. Chem. Soc. Dalton Trans. 1997, 4377–4384. [Google Scholar] [CrossRef]
- Ramakrishna, T.V.V.; Elias, A.J.; Vij, A. Synthesis and reactions of the ferrocene derived hydroxymethyl phosphine FcCH(CH3)P(CH2OH)2 and its sulfide: Crystal structures of [FcCH(CH3)P(S)R2] R=CH2OH, CH2CH2CN and FcCH(CH3)P(S)(CH2O)2PPh (Fc=ferrocenyl). J. Organomet. Chem. 2000, 602, 125–132. [Google Scholar] [CrossRef]
- Gonschorowsky, M.; Merz, K.; Driess, M. Cyclohexylbis(hydroxymethyl)phosphane: A hydrophilic phosphane capable of forming novel hydrogen-bonding networks. Eur. J. Inorg. Chem. 2006, 455–463. [Google Scholar] [CrossRef]
- Goodwin, N.J.; Henderson, W.; Nicholson, B.K. Hydrogen bonding in hydroxymethylphosphine chalcogenides. Inorg. Chim. Acta 2002, 335, 113–118. [Google Scholar] [CrossRef]
- Griffiths, D.V.; Groombridge, H.J.; Salt, M.C. Investigations into the oxidative stability of hydroxymethyl- and bis(hydroxymethyl)-phosphines. Phosphorus, Sulfur, Silicon, Relat. Elem. 2008, 183, 473–478. [Google Scholar] [CrossRef]
- Hellmann, H.; Bader, J.; Birkner, H.; Schumacher, O. Hydroxymethylphosphines, hydroxymethylphosphonium salts, and chloromethylphosphonium salts. Justus Liebigs Ann. Chem. 1962, 659, 49–63. [Google Scholar] [CrossRef]
- Fawcett, J.; Hoye, P.A.T.; Kemmitt, R.D.W.; Law, D.J.; Russell, D.R. Synthesis of bis(phosphinomethyl)amines via bis(hydroxymethyl)phosphonium salts. Isolation of 9,9-bis(hydroxymethyl)-9-phosphoniabicyclo[3.3.1]nonane hydrogen sulfate and chloride salts, and the crystal structures of [PPh2(CH2OH)2]+ Cl− and [(C6H11)2PCH2]2NCHMePh. J. Chem. Soc. Dalton Trans. 1993, 2563–2568. [Google Scholar] [CrossRef]
- Zingaro, R.A. Phosphine sulfides and selenides: The phosphorus-sulfur and phosphorus-selenium stretching frequencies. Inorg. Chem. 1963, 2, 192–196. [Google Scholar] [CrossRef]
- Tan, S.L.; Jotani, M.M.; Tiekink, E.R.T. Utilizing Hirshfeld surface calculations, non-covalent interaction (NCI) plots and the calculation of interaction energies in the analysis of molecular packing. Acta Crystallogr. E 2019, 75, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.J.; Mckinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer; v17; The University of Western Australia: Crawley, Australia, 2017. [Google Scholar]
- Strohalm, M.; Hassman, M.; Košata, B.; Kodíček, M. mMass data miner: An open source alternative for mass spectrometric data analysis. Rapid Commun. Mass Spectrom. 2008, 22, 905–908. [Google Scholar] [CrossRef] [PubMed]
- Rigaku Oxford Diffraction. CrysAlis PRO.; Rigaku Corporation: Oxford, UK, 2017. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Brandenburg, K.; Putz, H. DIAMOND.; Crystal Impact GbR: Bonn, Germany, 2006. [Google Scholar]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Henderson, W.; Okpareke, O.C.; Azizan, A.H.S.; Tiekink, E.R.T. [Dicyclohexyl(sulfanylidene)-λ5-phosphanyl]methanol. Molbank 2019, 2019, M1069. https://doi.org/10.3390/M1069
Henderson W, Okpareke OC, Azizan AHS, Tiekink ERT. [Dicyclohexyl(sulfanylidene)-λ5-phosphanyl]methanol. Molbank. 2019; 2019(3):M1069. https://doi.org/10.3390/M1069
Chicago/Turabian StyleHenderson, William, Obinna C. Okpareke, Ainnul Hamidah Syahadah Azizan, and Edward R. T. Tiekink. 2019. "[Dicyclohexyl(sulfanylidene)-λ5-phosphanyl]methanol" Molbank 2019, no. 3: M1069. https://doi.org/10.3390/M1069
APA StyleHenderson, W., Okpareke, O. C., Azizan, A. H. S., & Tiekink, E. R. T. (2019). [Dicyclohexyl(sulfanylidene)-λ5-phosphanyl]methanol. Molbank, 2019(3), M1069. https://doi.org/10.3390/M1069