6R/S-deutero-α-d-mannopyranoside 1-phosphate


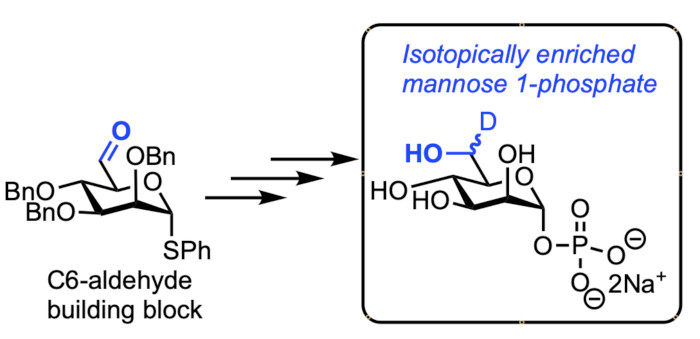
Abstract

{kind=link}
{kind=link}
1. Introduction
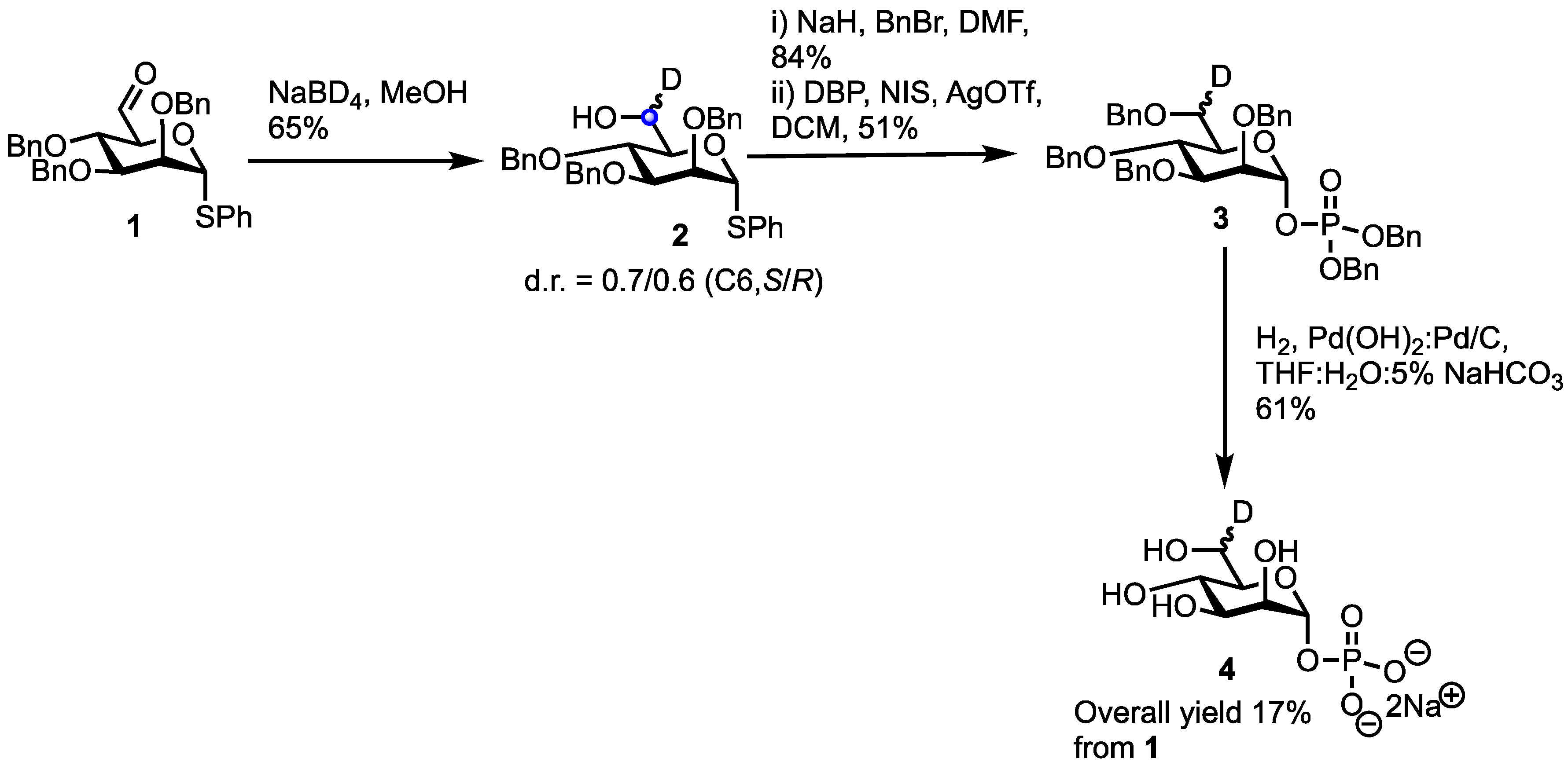
2. Results
3. Materials and Methods
3.1. General
3.2. Phenyl 2,3,4-tri-O-benzyl-6R/S-deutero-1-thio-α-d-mannopyranoside (2)
3.3. Dibenzyl (2,3,4,6-tetra-O-benzyl-6R/S-deutero-1-phosphate-α-d-mannopyranoside) (3)
3.4. R/S-deutero-α-d-mannopyranoside-1-phosphate (4)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Varki, A. Essentials of Glycobiology; Cold Spring Harbor: New York, NY, USA, 1999. [Google Scholar]
- Pale, P.; Whitesides, G.M. Synthesis of Glycosyl Phosphates Using the Fraser-Reid Activation. J. Org. Chem. 1991, 56, 4547–4549. [Google Scholar] [CrossRef]
- Fujita, S.; Oka, N.; Matsumura, F.; Wada, T. Synthesis of Oligo (α-D-glycosyl phosphate) Derivatives by a Phosphoramidite Method via Boranophosphate Intermediates. J. Org. Chem. 2011, 76, 2648–2659. [Google Scholar] [CrossRef] [PubMed]
- Shashkov, A.S.; Streshinskaya, G.M.; Tulskaya, E.M.; Senchenkova, S.N.; Baryshnikova, L.M.; Dmitrenok, A.S.; Ostash, B.E.; Fedorenko, V.A. Cell wall glycopolymers of Streptomyces albus, Streptomyces albidoflavus and Streptomyces pathocidini. Int. J. Gen. Mol. Microbiol. 2016, 109, 923–936. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Ma, M.; Wang, L.; Zhang, Y.; Jiang, R.; Rong, B.; Li, Y. Biochemical characterization of a novel bifunctional glycosyl-1-phosphate transferase involved in the exopolysaccharide biosynthesis. Biochem. Biophys. Res. Commun. 2015, 465, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Ahmadipour, S.; Miller, G.J. Recent advances in the chemical synthesis of sugar-nucleotides. Carbohydr. Res. 2017, 451, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Ahmadipour, S.; Beswick, L.; Miller, G.J. Recent advances in the enzymatic synthesis of sugar-nucleotides using nucleotidylyltransferases and glycosyltransferases. Carbohydr. Res. 2018, 469, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Hoffmeister, D.; Yang, J.; Liu, L.; Thorson, J.S. Creation of the first anomeric D/L-sugar kinase by means of directed evolution. Proc. Natl. Acad. Sci. USA 2003, 100, 13184–13189. [Google Scholar] [CrossRef] [PubMed]
- Pergolizzi, G.; Kuhaudomlarp, S.; Kalita, E.; Field, R.A. Glycan Phosphorylases in Multi-Enzyme Synthetic Processes. Protein Pept. Lett. 2017, 24, 696–709. [Google Scholar] [CrossRef] [PubMed]
- Nakai, H.; Kitaoka, M.; Svensson, B.; Ohtsubo, K. Recent development of phosphorylases possessing large potential for oligosaccharide synthesis. Curr. Opin. Chem. Biol. 2013, 17, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Auriol, D.; Lefevre, F.; Nalin, R.; Redziniak, G. Cosmetic and pharmaceutical composition comprising N-acetylglucosamine-6-phosphate. US 20130012475, 1 April 2011. [Google Scholar]
- Woodyer, R.; Taylor, P.; Demirjian, D. Activated Sugars. US 20140024082A1, 2011. [Google Scholar]
- De Groeve, M.R.; Depreitere, V.; Desmet, T.; Soetaert, W. Enzymatic production of a-D-galactose 1-phosphate by lactose phosphorolysis. Biotechnol. Lett. 2009, 31, 1873–1877. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Huang, K.; Wei, M.; Meisner, J.; Liu, Y.; Garner, K.; Zang, L.; Wang, X.; Li, X.; Fang, J.; et al. Facile Enzymatic Synthesis of Ketoses. Angew. Chem. Int. Ed. 2015, 54, 12654–12658. [Google Scholar] [CrossRef] [PubMed]
- Kakinuma, K. Efficient and mild lactonization method for the synthesis of macrolides. J. Am. Chem. Soc. 1981, 103, 5614–5616. [Google Scholar] [CrossRef]
- Dong, H.; Mahmud, T.; Tornus, I.; Lee, S.; Floss, H.G. Biosynthesis of the Validamycins: Identification of Intermediates in the Biosynthesis of Validamycin A by Streptomyces hygroscopicus var. l imoneus. J. Am. Chem. Soc. 2001, 123, 2733–2742. [Google Scholar] [CrossRef]
- Xu, L.; Price, N.P.J. Stereoselective synthesis of chirally deuterated (S)- D-(6-2H1) glucose. Carbohydr. Res. 2004, 339, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Read, J.A.; Ahmed, R.A.; Tanner, M.E. Efficient Chemoenzymatic Synthesis of ADP-d-glycero-β-D-manno-Heptose and a Mechanistic Study of ADP-l-glycero-d-manno-Heptose 6-Epimerase. Org. Lett. 2005, 7, 2457–2460. [Google Scholar] [CrossRef]
- Ahmadipour, S.; Pergolizzi, G.; Rejzek, M.; Field, R.A.; Miller, G.J. Chemoenzymatic Synthesis of C6-Modified Sugar Nucleotides To Probe the GDP-D-Mannose Dehydrogenase from Pseudomonas aeruginosa. Org. Lett. 2019, 21, 4415–4419. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmadipour, S.; Miller, G.J. 6R/S-deutero-α-d-mannopyranoside 1-phosphate. Molbank 2019, 2019, M1068. https://doi.org/10.3390/M1068
Ahmadipour S, Miller GJ. 6R/S-deutero-α-d-mannopyranoside 1-phosphate. Molbank. 2019; 2019(3):M1068. https://doi.org/10.3390/M1068
Chicago/Turabian StyleAhmadipour, Sanaz, and Gavin J. Miller. 2019. "6R/S-deutero-α-d-mannopyranoside 1-phosphate" Molbank 2019, no. 3: M1068. https://doi.org/10.3390/M1068
APA StyleAhmadipour, S., & Miller, G. J. (2019). 6R/S-deutero-α-d-mannopyranoside 1-phosphate. Molbank, 2019(3), M1068. https://doi.org/10.3390/M1068