(2S,5R)-2-Isopropyl-5-methylcyclohexanone Hydrazones

 ,
,  , and
, and 
Abstract
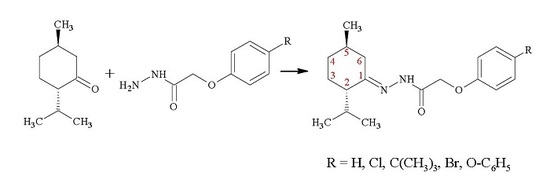
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
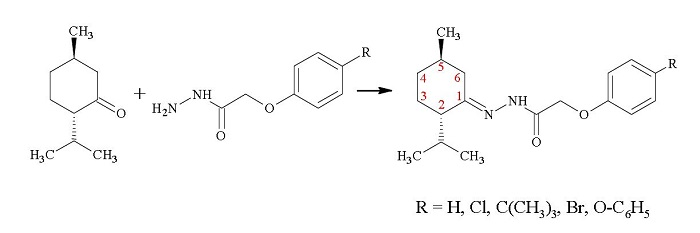
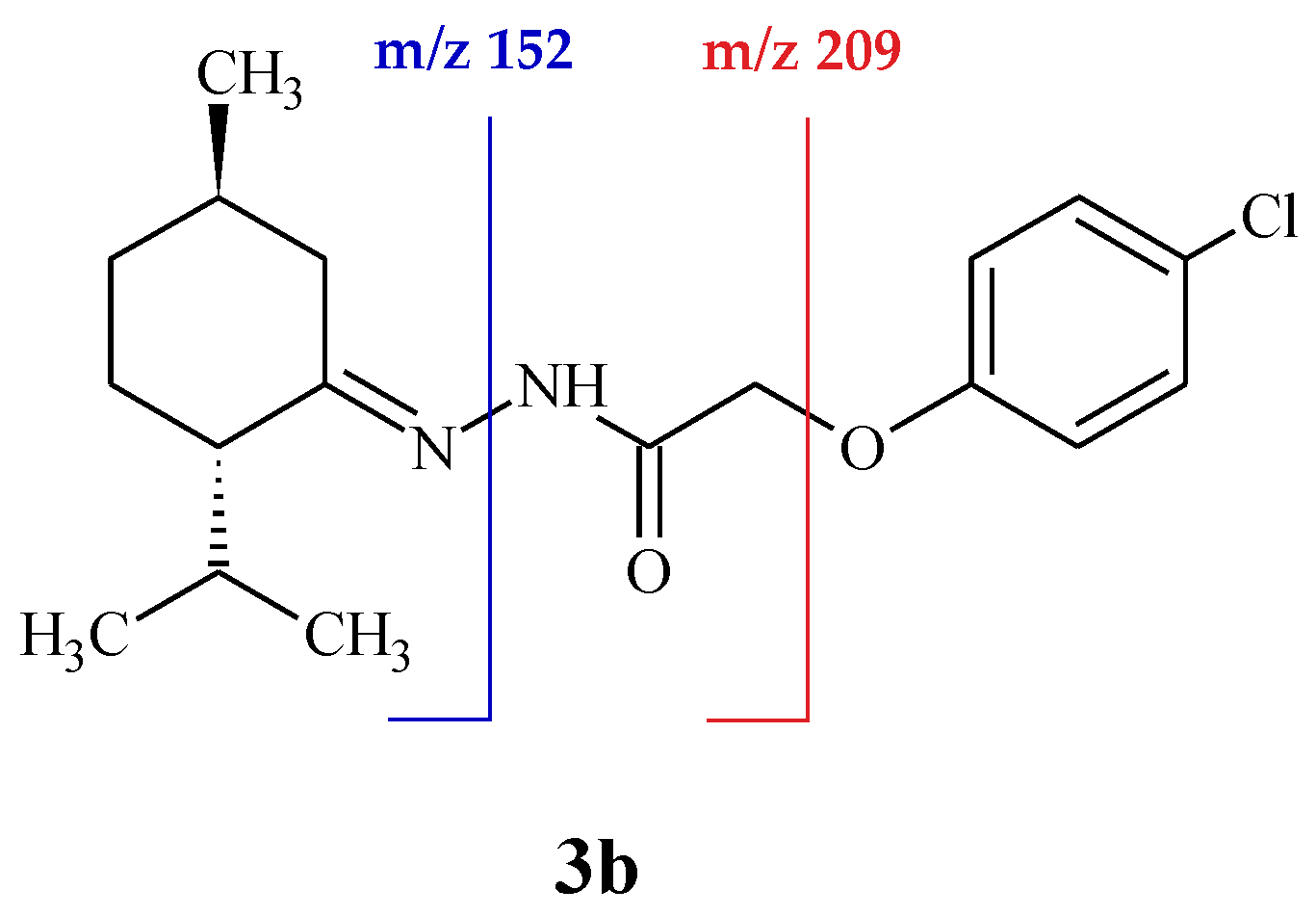
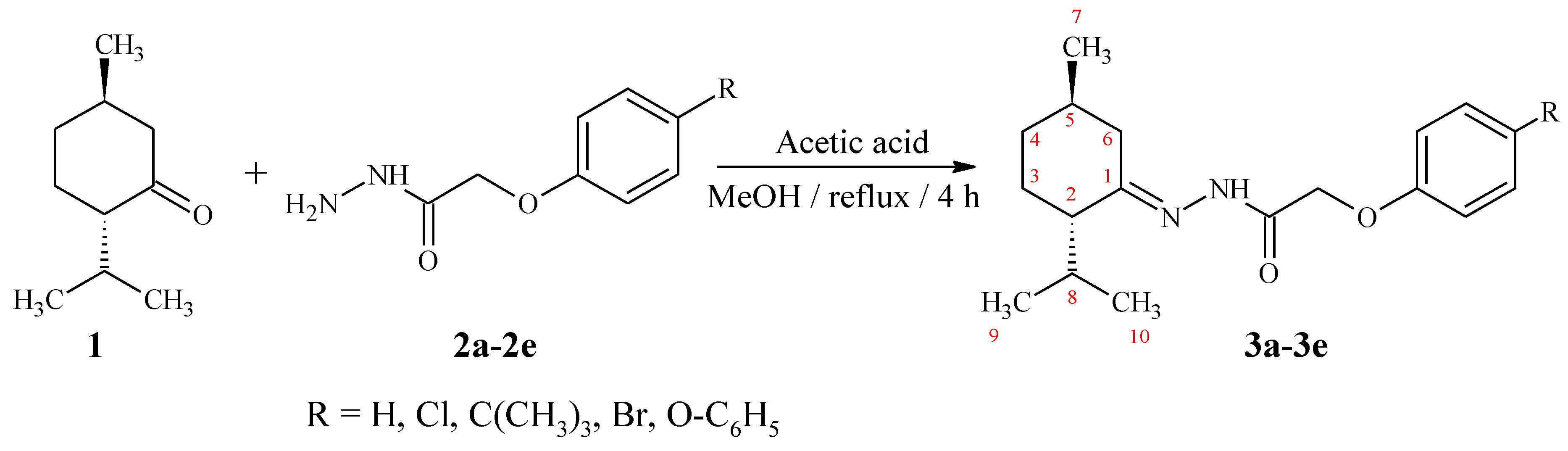
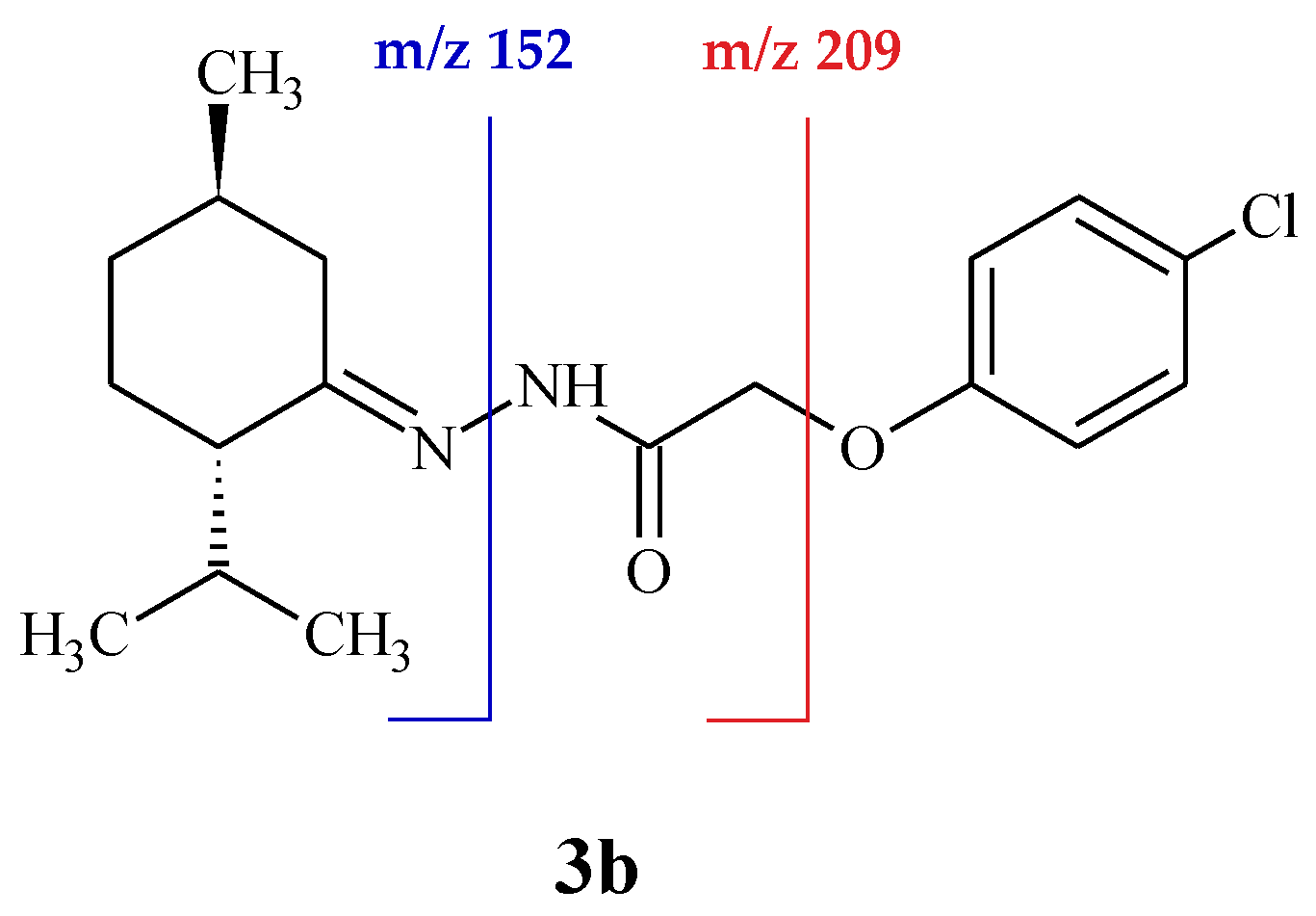
2. Results and Discussion
3. Materials and Methods
3.1. General Information
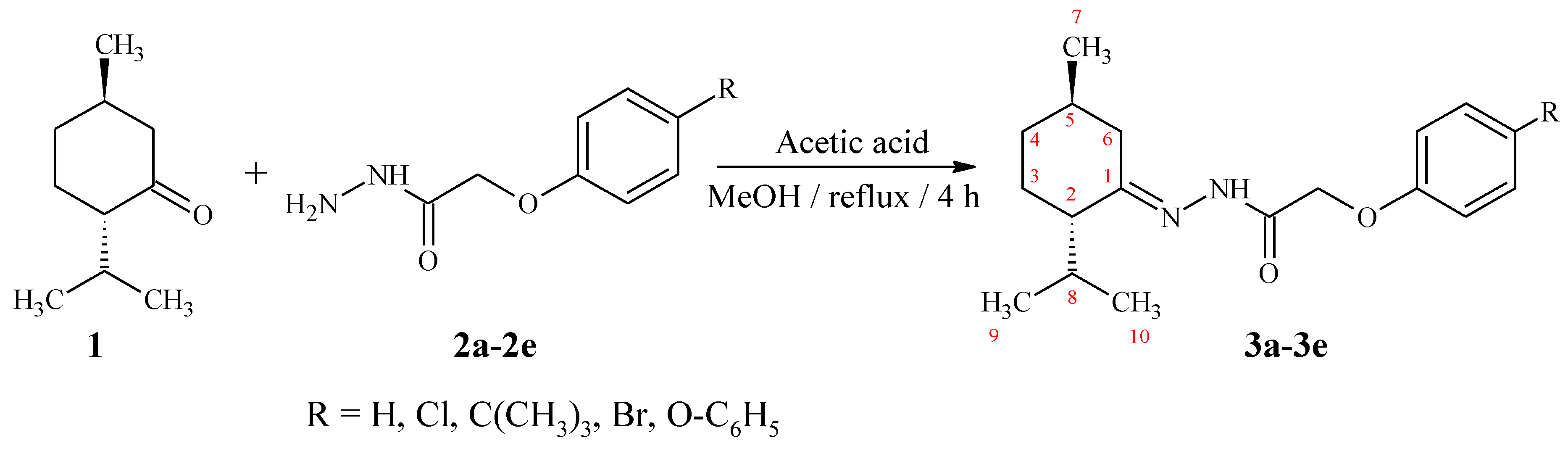
3.2. Synthesis of (2S,5R)-2-Isopropyl-5-methylcyclohexanone hydrazones (3a–3e)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nesterkina, M.; Shishkina, S.; Maltsev, G.; Rakipov, I.; Kravchenko, I. (1R,2S,5R)-2-Isopropyl-5-methylcyclohexyl 4-aminobutyrate hydrochloride. Molbank 2017, 2017, M956. [Google Scholar] [CrossRef]
- Nesterkina, M.; Kravchenko, I. Synthesis and pharmacological properties of novel esters based on monocyclic terpenes and GABA. Pharmaceuticals 2016, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Vriens, J.; Nilius, B.; Vennekens, R. Herbal compounds and toxins modulating TRP channels. Curr. Neuropharmacol. 2008, 6, 79–96. [Google Scholar] [PubMed]
- Sánchez-Borzone, M.E.; Marin, L.D.; García, D.A. Effects of insecticidal ketones present in mint plants on GABAA receptor from mammalian neurons. Pharmacogn. Mag. 2017, 13, 114–117. [Google Scholar] [PubMed]
- Turan-Zitouni, G.; Yurttaş, L.; Kaplancıklı, Z.A.; Can, Ö.D.; Özkay, Ü.D. Synthesis and anti-nociceptive, anti-inflammatory activities of new aroyl propionic acid derivatives including N-acylhydrazone motif. Med. Chem. Res. 2015, 24, 2406–2416. [Google Scholar] [CrossRef]
- Pages, N.; Maurois, P.; Bac, P.; Eynde, J.J.V.; Tamariz, J.; Labarrios, F.; Chamorro, G.; Vamecq, J. The α-asarone/clofibrate hybrid compound, 2-methoxy-4-(2-propenyl)phenoxyacetic acid (MPPA), is endowed with neuroprotective and anticonvulsant potentialities. Biomed. Aging Pathol. 2011, 1, 210–215. [Google Scholar] [CrossRef]
- Rollas, S.; Küçükgüzel, S.G. Biological activities of hydrazone derivatives. Molecules 2007, 12, 1910–1939. [Google Scholar] [CrossRef] [PubMed]
- Palla, G.; Predieri, G.; Domiano, P.; Vignali, C.; Turner, W. Conformational behaviour and E/Z isomerization of N-acyl and N-aroylhydrazones. Tetrahedron 1986, 42, 3649–3654. [Google Scholar] [CrossRef]
- Johansson, A.; Kollman, P.; Rothenberg, S.; McKelvey, J. Hydrogen bonding ability of the amide group. J. Am. Chem. Soc. 1974, 96, 3794–3800. [Google Scholar] [CrossRef]
- Kaur, P.; Kaur, R.; Goswami, M. A review on methods of synthesis of 1,2,4-triazole derivatives. IRJP 2018, 9, 1–35. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nesterkina, M.; Barbalat, D.; Zheltvay, I.; Rakipov, I.; Atakay, M.; Salih, B.; Kravchenko, I. (2S,5R)-2-Isopropyl-5-methylcyclohexanone Hydrazones. Molbank 2019, 2019, M1062. https://doi.org/10.3390/M1062
Nesterkina M, Barbalat D, Zheltvay I, Rakipov I, Atakay M, Salih B, Kravchenko I. (2S,5R)-2-Isopropyl-5-methylcyclohexanone Hydrazones. Molbank. 2019; 2019(2):M1062. https://doi.org/10.3390/M1062
Chicago/Turabian StyleNesterkina, Mariia, Dmytro Barbalat, Ivan Zheltvay, Ildar Rakipov, Mehmet Atakay, Bekir Salih, and Iryna Kravchenko. 2019. "(2S,5R)-2-Isopropyl-5-methylcyclohexanone Hydrazones" Molbank 2019, no. 2: M1062. https://doi.org/10.3390/M1062
APA StyleNesterkina, M., Barbalat, D., Zheltvay, I., Rakipov, I., Atakay, M., Salih, B., & Kravchenko, I. (2019). (2S,5R)-2-Isopropyl-5-methylcyclohexanone Hydrazones. Molbank, 2019(2), M1062. https://doi.org/10.3390/M1062