Abstract
The synthesis, spectroscopic characterization and X-ray crystal structure of the title compound, (4-tolyl)3PAu[SC(O-i-Pr)=NC6H4NO2-4] (1) are described. Spectroscopy exhibited the expected features confirming the formation of the compound. The molecular structure of 1 confirms the expected linear P–Au–S coordination geometry defined by thiolate-S and phosphane-P atoms. The nearly 7° deviation from linearity is ascribed to the close approach of the imine-bound phenyl group, indicative of a semi-localized Au…π(arene) interaction. The three-dimensional molecular packing is consolidated by methyl- and tolyl-C–H…O(nitro) and tolyl-C–H…π(tolyl) interactions.
1. Introduction
Gold(I) is well known to be a carbophilic Lewis acid and this characteristic underscores the great current interest of “catalytic gold” [1,2]. This carbophilicity extends to the solid-state with Au…π(arene) interactions shown to sustain well-defined supramolecular aggregation patterns that occur independently of other recognizable supramolecular synthons, such as hydrogen bonding [3,4,5]. The energy associated with Au…π(arene) interactions has been calculated to be around 12 kcal/mol [6] which is of the same order of magnitude as both conventional hydrogen bonding and aurophilic interactions [7,8]. Herein, the crystal and molecular structures of (4-tolyl)3PAu[SC(O-i-Pr)=NC6H4NO2-4] (1), Scheme 1, is described along with spectroscopic (i.e., 1H, 13C{1H} and 31P{1H} NMR, UV and IR) data. As discussed below, the molecule of 1 features an intramolecular Au…π(arene) interaction whereas many of the literature analogues are known to present Au…O interactions instead [9], a conformation achieved by a rotation about the C–S bond, as indicated by the green arrow in Scheme 1. Calculations have shown that for the crystallographically characterized conformational polymorphs of Ph3PAu[SC(OEt)=NPh], the form with the Au…π(arene) interaction [9] is over 5 kcal/mol more stable than the form featuring the intramolecular Au…O interaction [10]. As a continuation of studies in this area [6,7,8,9,10], the structural characterization of 1 is described herein.
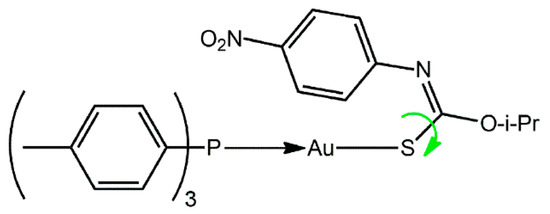
Scheme 1.
Chemical diagram for 1. The green arrow indicates the possibility of rotation about the C–S bond to place the alkoxide-oxygen atom in proximity to the gold(I) center rather than the nitrophenyl ring.
2. Results and Discussion
Compound 1 was prepared in good yield (79%) by standard methods [11] and was characterized by spectroscopy; see Supplementary Materials for original spectra. The NMR data showed the expected resonances and integration (1H). In the IR, characteristic bands ascribed to ν(C=N) [1330 (vs)], ν(C–O) [1139 (vs)] and ν(C–S) [1099 (vs)] were observed. No evidence was found for a signal due to N–H in the 1H-NMR spectrum or of a band due to ν(N–H) in the IR spectrum [12]. In the UV spectrum, the most prominent band at λabs 240 nm is attributed to the thiolate ligand-centered π→π* transition [13]. The two shoulders at 267 and 276 nm are assigned to intra-ligand transition of tri(4-tolyl)phosphane, while the band at 340 nm arises from the 1[n(S)→π*(C6H4NO2)] intra-ligand charge transfer [13]. Full characterization of 1 was achieved by single-crystal X-ray crystallography.
The molecular structure of 1 is shown in Figure 1 with salient geometric parameters included in the figure caption. To a first approximation, the gold(I) center is linearly coordinated by thiolate-S1 and phosphane-P1 atoms. The thiolate assignment is readily vindicated by comparing the C1–S1 and C1–N1 bond lengths of 1, i.e., 1.771(5) and 1.260(8) Å, respectively, with the equivalent bonds of recorded for the free ligand, i.e., S=C(O-i-Pr)N(H)C6H4NO2-4, of 1.678 (3) and 1.352 (3) Å, respectively [12]. This shows considerable lengthening and shortening in the thiocarbamate anion consistent with significant reorganization of π-electron density in 1 compared to that seen in the original thiocarbamide molecule [12]. The systematic changes in the angles subtended at the quaternary-C1 atom are also consistent with above, with the widest angles subtended by the imine-N1 atom, e.g., S1‒C1‒O1 is the narrowest angle in 1, at 108.3(4)°, but is the widest, at 126.2(2)°, in the free molecule [12]. The central CNOS chromophore is strictly planar with the r.m.s. deviation of the fitted atoms being 0.0082 Å with the maximum deviation from this plane being 0.014(4) Å, for the C1 atom. There is an almost orthogonal relationship between this plane and the appended phenyl ring with the dihedral angle computing to 85.80(15)°. A small twist between the phenyl ring and the 4-nitro group connected to it is evident, as seen in the dihedral angle between the planes of 7.4(8)°.
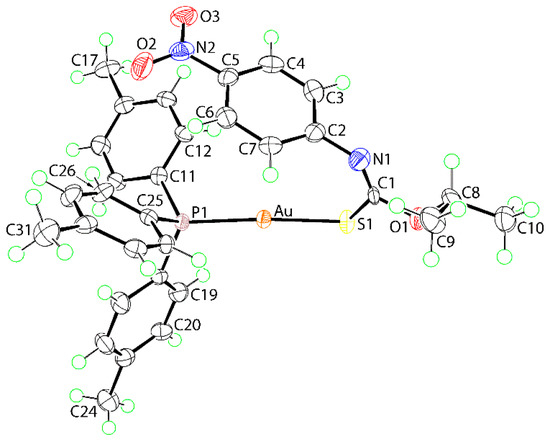
Figure 1.
The molecular structure of 1 showing atom labelling and displacement ellipsoids at the 70% probability level. Selected geometric parameters: Au‒S1 = 2.2993(12), Au‒P1 = 2.2649(12), C1‒S1 = 1.771(5), C1‒O1 = 1.348(6), C1=N1 = 1.260(8) Å; Au‒S1‒C1 = 107.33(16), S1‒C1‒O1 = 108.3(4), S1‒C1‒N1 = 131.1(4), O1‒C1‒N1 = 120.5(5)°.
A key feature of the structure is found in the relative orientation of the N1-bound phenyl ring which is disposed over the gold(I) center. The Au…ring centroid(C2-C7) distance is 3.50 Å with the shortest separations found for Au…C7 and Au…C2 of 3.173(6) and 3.244(5) Å, suggesting the nature of the interaction is semi-localized [14,15] towards the C2–C7 bond. The specific Au…C interactions are less than the sum of the van der Waals radii of gold and carbon, i.e., 3.36 Å [16] and the Au…ring centroid(C2–C7) distance is less than the sum of the Waals radii of gold and a phenyl ring (taken as 1.9 Å [17]) i.e., 3.56 Å. The relatively close proximity of the phenyl ring to gold is likely to be responsible for the deviation of the S1-Au-P1 angle, i.e., 173.15(4)°, from ideal linearity.
In the absence of conventional hydrogen bonding interactions, the molecular packing in the crystal of 1 features non-covalent interactions of type C‒H…O and C‒H…π, based on the distance criteria embodied in PLATON [18]; geometric parameters characterizing these are listed in Table 1.

Table 1.
Geometric parameters (Å, °) characterizing the non-covalent (A–H···B) interactions present in the crystal of 1.
Each of the nitro-oxygen atoms participates in a C‒H…O interaction, with the donor atoms being methyl-H and tolyl-H. The other interaction identified is an end-on tolyl-C‒H…π(tolyl) contact. These interactions extend in three-dimensions to consolidate the crystal. A view of the unit cell contents is shown in Figure 2.
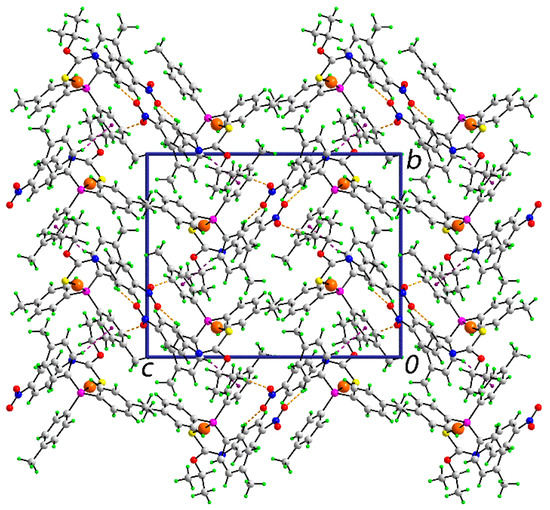
Figure 2.
Molecular packing in the crystal of 1: a view of the unit cell contents in projection down the a-axis. The C‒H…O and C‒H…π interactions are shown as orange and purple dashed lines, respectively.
Until the publication of a systematic analysis of R3PAu[SC(OMe)=NR’] structures, for R, R’ = aryl [11], all molecules with that general formula had featured intramolecular Au…O interactions. However, with the judicious choice of R and R’, e.g., isomeric tolyl groups, intramolecular Au…π(arene) interactions could be promoted. For example, when R = 4-tolyl, Au…π interactions formed, unless precluded by steric crowding. Subsequent studies, in particular, based on computational chemistry [6,9], suggest that molecules having intramolecular Au…π interactions are thermodynamically favored.
In conclusion, the X-ray crystal structure determination of (4-tolyl)3PAu[SC(O-i-Pr)=NC6H4NO2-4] confirms the spectroscopy and discloses an intramolecular, semi-localized Au…π interaction, which is expected based on electronic, steric, and thermodynamic grounds.
3. Materials and Methods
3.1. General Information
All standard chemicals and solvents were sourced from Merck (Darmstadt, Germany) and used without further purification. The melting point was determined on a Biobase automatic melting point apparatus MP450 (Biobase Group, Jinan, Shandong Province, China). The IR spectrum was measured on a Bruker Vertex 70v FTIR (Billerica, MA, USA) spectrophotometer from 4000 to 400 cm−1. Elemental analyses were performed on a Leco TruSpec Micro CHN Elemental Analyser (Saint Joseph, MI, USA). 1H and 13C{1H} NMR spectra were recorded in CDCl3 solution on a Bruker Ascend 400 MHz NMR (Billerica, MA, USA) spectrometer with chemical shifts relative to tetramethylsilane. 31P{1H} NMR spectra were recorded in CDCl3 solution on the same instrument but with the chemical shifts recorded relative to 85% aqueous H3PO4 as the external reference. The optical absorption spectra were obtained from an acetonitrile solution of 1 × 10−5 M in the range 200–800 nm on a Shimadzu UV-3600 plus UV/VIS/NIR (Shimadzu Corporation, Kyoto Prefecture, Japan) spectrophotometer.
3.2. Synthesis and Characterization
NaOH (0.2 mmol, 0.008 g) in MeOH (1 mL) was added to a suspension of tri(4-tolyl)phosphanegold(I) chloride [11] (0.2 mmol, 0.107 g) in MeOH (5 mL), followed by the addition of O-isopropyl-N-4-nitrophenyl thiocarbamide [12] (0.2 mmol, 0.048 g) in MeOH (5 mL). The resulting mixture was stirred at 50 °C for 2 h and then left for slow evaporation under ambient conditions. Yellow crystals were collected after two weeks. Yield: 0.117 g, 79%. M. pt: 143.0–143.7 °C. Anal. Calc. for C31H32AuN2O3PS: C, 50.28; H, 4.36; N, 3.78. Found: C, 50.59; H, 4.32; N, 3.50. IR (cm−1): 1501 (s), 1309 (s) ν(NO2), 1330 (vs) ν(C=N), 1139 (vs) ν(C-O), 1099 (vs) ν(C-S). 1H NMR (CDCl3): δ 7.92 (d, 2H, 3-phenyl-H, 3JHH = 8.96 Hz), 7.31–7.23 (m, br, 12H, 4-tolyl), 6.89 (d, 2H, 2-phenyl-H, 3JHH = 8.96 Hz), 5.24 (sept, 1H, OCH, 3JHH = 6.20 Hz), 2.41 (s, 9H, tolyl-CH3), 1.34 (d, 6H, i-Pr-CH3, 3JHH = 6.20 Hz) ppm. 13C{1H} NMR (CDCl3): δ 164.9 (Cq), 157.7 (Ph, C1), 142.6 (Ph, C4), 142.3 (d, 4-tolyl, 4JPC = 2.31 Hz), 133.9 (d, 3-tolyl, 3JPC = 14.12 Hz), 129.9 (d, 2-tolyl, 2JCP = 11.90 Hz), 126.2 (d, 1-tolyl, 1JPC = 60.02 Hz), 124.9 (Ph, C3), 122.5 (Ph, C2), 71.4 (OCH), 22.0 (i-Pr-CH3), 21.5 (d, tolyl-CH3, 5JPC = 1.04 Hz) ppm. 31P{1H} NMR (CDCl3): δ 36.7 ppm. UV (acetonitrile; nm, L·cm−1·mol−1): λabs = 240, ε = 42,500; λabs(sh) = 267, ε = 13,400; λabs(sh) = 276, ε = 8800; λabs = 340, ε = 14,500.
3.3. X-ray Crystallography
A SuperNova Dual AtlasS2 diffractometer fitted with Cu Kα radiation (λ = 1.54178 Å) was employed for the intensity data collection at T = 100 K for 1. CrysAlis Pro [19] was employed for data reduction and absorption correction. Of the 20549 reflections included up to θmax = 67.1°, 5276 were unique (Rint = 0.044) and of these, 5133 data satisfied the I ≥ 2σ(I) criterion. The structure was solved by direct methods [20] and refined (anisotropic displacement parameters and C-bound H atoms in the riding model approximation) on F2 [21]. A weighting scheme of the form w = 1/[σ2(Fo2) + (0.017P)2] was introduced, where P = (Fo2 + 2Fc2)/3). Based on the refinement of 357 parameters, the final values of R (obs. data) and wR (all data) were 0.019 and 0.044, respectively. The absolute structure was determined based on differences in Friedel pairs included in the data set. The molecular structure diagram was generated with ORTEP for Windows [22] and the packing diagram using DIAMOND [23].
Crystal data for C31H32AuN2O3PS (1): M = 740.58, orthorhombic, P212121, a = 11.19860(10), b = 14.59850(10), c = 18.2307(2) Å, V = 2980.41(5) Å3, Z = 4, Dx = 1.650 g cm−3, F(000) = 1464, µ = 10.712 mm−1.
Supplementary Materials
The following are available online. 1H, 13C{1H} and 31P{1H} NMR, UV and IR spectra, and crystallographic data for 1 in Crystallographic Information File (CIF) format. CCDC 1874265 also contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html.
Author Contributions
C.I.Y. was the only experimentalist, who obtained and analysed all data, apart from the X-ray crystallography, which was performed by E.R.T.T.
Funding
This research received no external funding. The APC was funded by Sunway University.
Acknowledgments
The X-ray crystallography laboratory at Sunway University is thanked for providing the X-ray intensity data.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Haruta, M. Gold as a novel catalyst in the 21st century: Preparation, working mechanism and applications. Gold Bull. 2004, 37, 27–36. [Google Scholar] [CrossRef]
- Rudolph, M.; Hashmi, A.S.K. Gold catalysis in total synthesis—An update. Chem. Soc. Rev. 2012, 41, 2448–2462. [Google Scholar] [CrossRef] [PubMed]
- Tiekink, E.R.T.; Zukerman-Schpector, J. Gold…π aryl interactions as supramolecular synthons. CrystEngComm 2009, 11, 1176–1186. [Google Scholar] [CrossRef]
- Tiekink, E.R.T.; Zukerman-Schpector, J. A structural survey of metal/π heteroaromatic supramolecular synthons for metal = tellurium, tin, and gold. CrystEngComm 2009, 11, 2701–2711. [Google Scholar] [CrossRef]
- Caracelli, I.; Zukerman-Schpector, J.; Tiekink, E.R.T. Supra-molecular synthons based on gold…π(arene) interactions. Gold Bull. 2013, 46, 81–89. [Google Scholar] [CrossRef]
- Yeo, C.I.; Khoo, C.-H.; Chu, W.-C.; Chen, B.-J.; Chu, P.-L.; Sim, J.-H.; Cheah, Y.-K.; Ahmad, J.; Halim, S.N.A.; Seng, H.-L.; et al. The importance of Au…π(aryl) interactions in the formation of spherical aggregates in binuclear phosphanegold(I) complexes of a bipodal thiocarbamate dianion: A combined crystallographic and computational study, and anti-microbial activity. RSC Adv. 2015, 5, 41401–41411. [Google Scholar] [CrossRef]
- Tiekink, E.R.T. Supramolecular assembly of molecular gold(I) compounds: An evaluation of the competition and complementarity between aurophilic (Au…Au) and conventional hydrogen bonding interactions. Coord. Chem. Rev. 2014, 275, 130–153. [Google Scholar] [CrossRef]
- Tiekink, E.R.T. Supramolecular assembly based on “emerging” intermolecular interactions of particular interest to coordination chemists. Coord. Chem. Rev. 2017, 345, 209–228. [Google Scholar] [CrossRef]
- Yeo, C.-I.; Tan, S.L.; Otero-de-la-Roza, A.; Tiekink, E.R.T. A conformational polymorph of Ph3PAu[SC(OEt)=NPh] featuring an intramolecular Au…π interaction. Z. Kristallogr. Cryst. Mater. 2016, 231, 653–661. [Google Scholar] [CrossRef]
- Hall, V.J.; Tiekink, E.R.T. Crystal structure of triphenylphosphine-(N-phenyl-O-ethylthiocarbamato)gold(I), (C6H5)3PAu(SC(=NPh)OEt). Z. Kristallogr. 1993, 203, 313–315. [Google Scholar] [CrossRef]
- Kuan, F.S.; Ho, S.Y.; Tadbuppa, P.P.; Tiekink, E.R.T. Electronic and steric control over Au…Au, C-H…O and C-H…interactions in the crystal structures of mononuclear triarylphosphinegold(I) carbonimidothioates: R3PAu[SC(OMe)=NR′] for R = Ph, o-tol, m-tol or p-tol, and R′ = Ph, o-tol, m-tol, p-tol or C6H4NO2-4. CrystEngComm 2008, 10, 548–564. [Google Scholar] [CrossRef]
- Ellis, C.A.; Tiekink, E.R.T.; Zukerman-Schpector, J. (E)-O-Isopropyl N-(4-nitrophenyl)thiocarbamate. Acta Crystallogr. E 2008, 64, o345. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.Y.; Cheng, E.C.-C.; Tiekink, E.R.T.; Yam, V.W.-W. Luminescent phosphine gold(I) thiolates: Correlation between crystal structure and photoluminescent properties in [R3PAu{SC(OMe)=NC6H4NO2-4}] (R = Et, Cy, Ph) and [(Ph2P-R-PPh2){AuSC(OMe)=NC6H4NO2-4}2] (R = CH2, (CH2)2, (CH2)3, (CH2)4, Fc). Inorg. Chem. 2006, 45, 8165–8174. [Google Scholar] [CrossRef] [PubMed]
- Schollmeyer, D.; Shishkin, O.V.; Rühl, T.; Vysotsky, M.O. OH–π and halogen–π interactions as driving forces in the crystal organisations of tri-bromo and tri-iodo trityl alcohols. CrystEngComm 2008, 10, 715–723. [Google Scholar] [CrossRef]
- Shishkin, O.V. Evaluation of true energy of halogen bonding in the crystals of halogen derivatives of trityl alcohol. Chem. Phys. Lett. 2008, 458, 96–100. [Google Scholar] [CrossRef]
- Bondi, A. Van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Janiak, J. A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands. J. Chem. Soc. Dalton Trans. 2000, 3885–3896. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Rigaku Oxford Diffraction, CrysAlis PRO; Agilent Technologies Inc.: Santa Clara, CA, USA, 2017.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Brandenburg, K. Diamond; Crystal Impact GbR: Bonn, Germany, 2006. [Google Scholar]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).