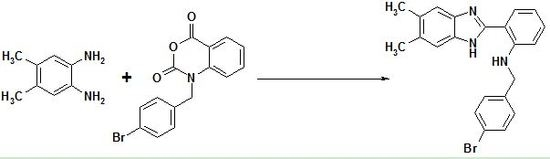
N-(4-Bromobenzyl)-2-(5,6-dimethyl-1H-benzo[d]imid-azol-2-yl)benzeneamine

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
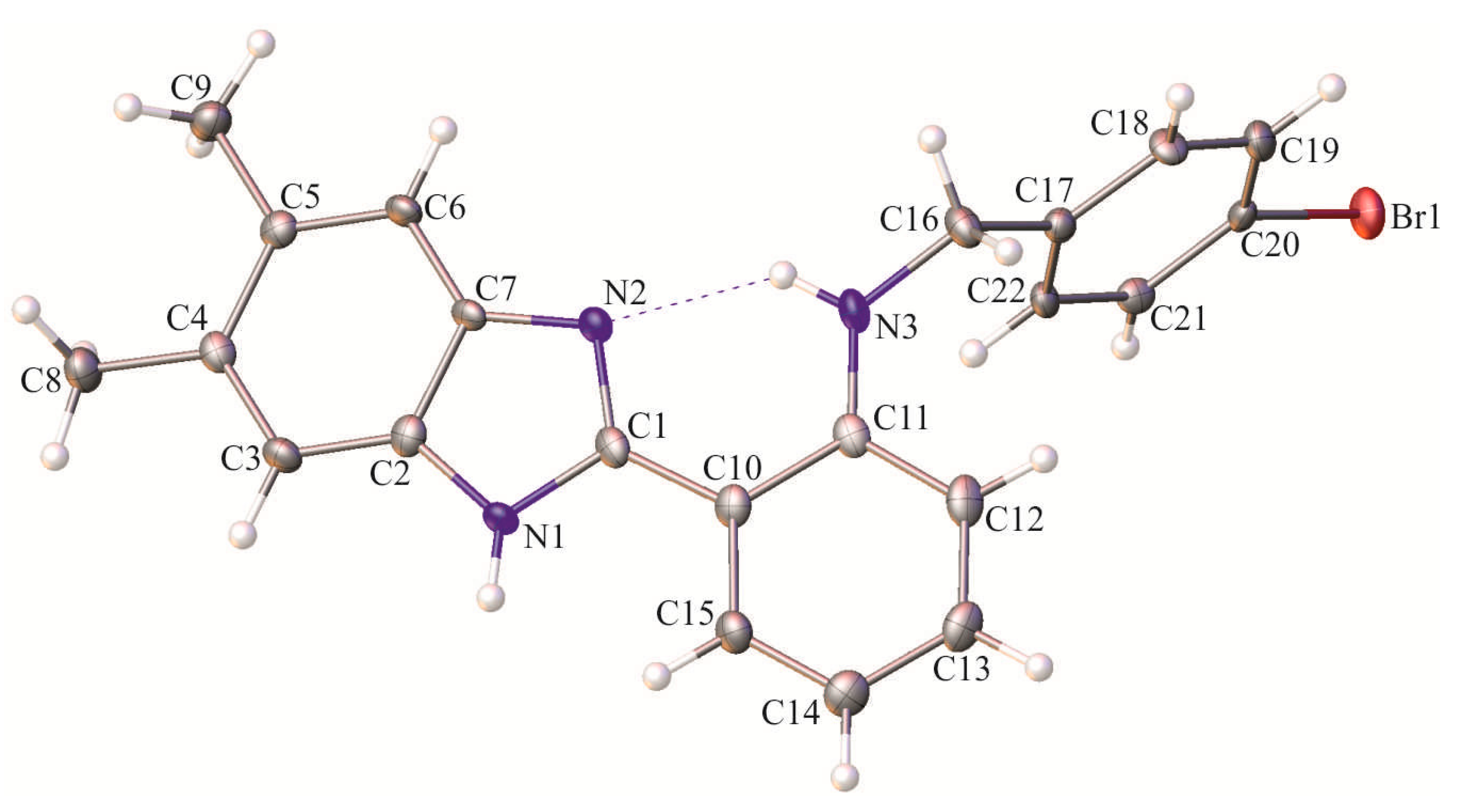
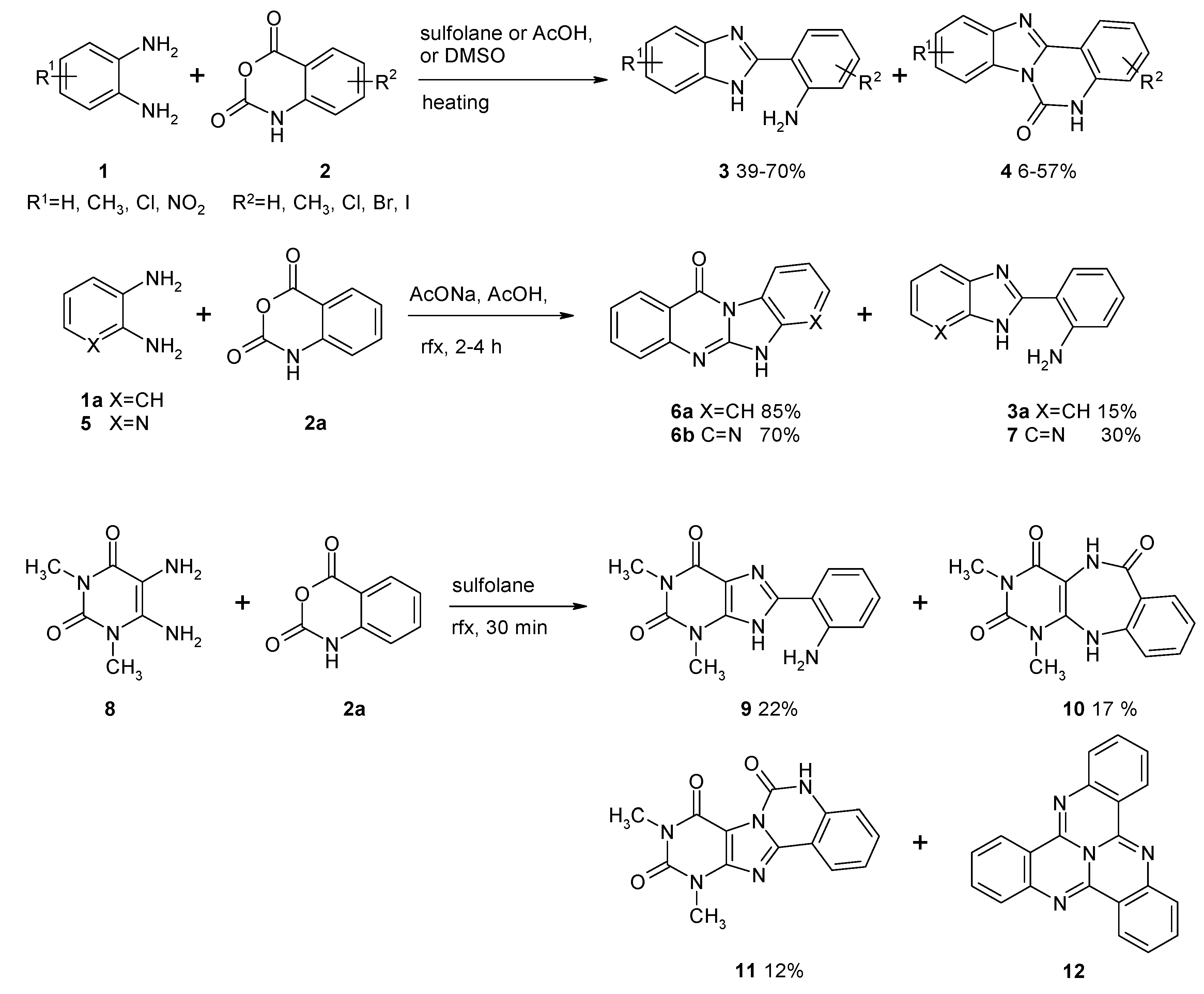
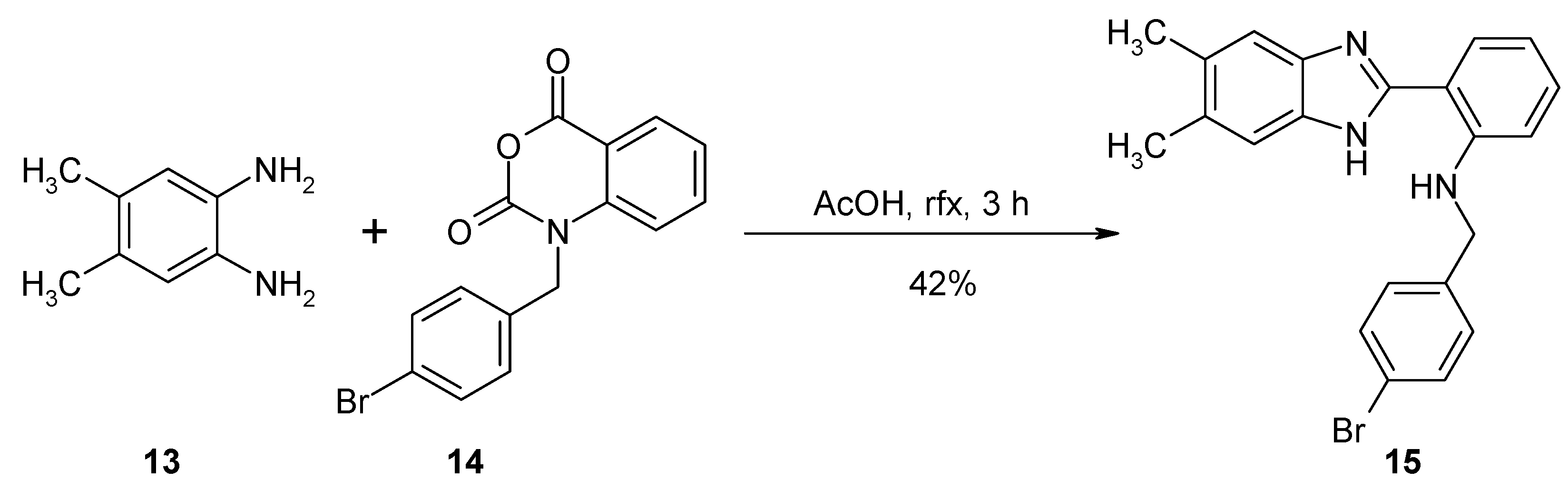
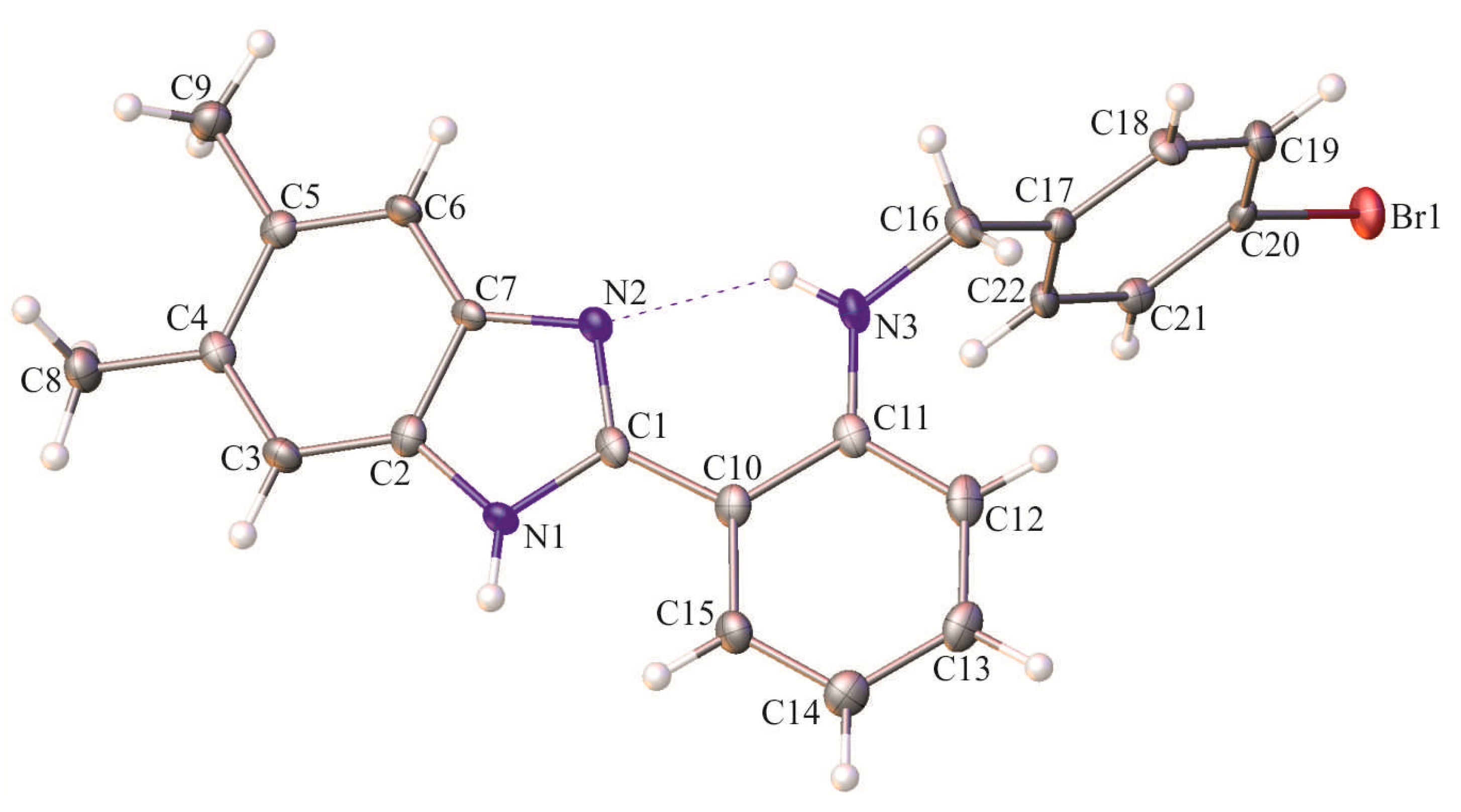
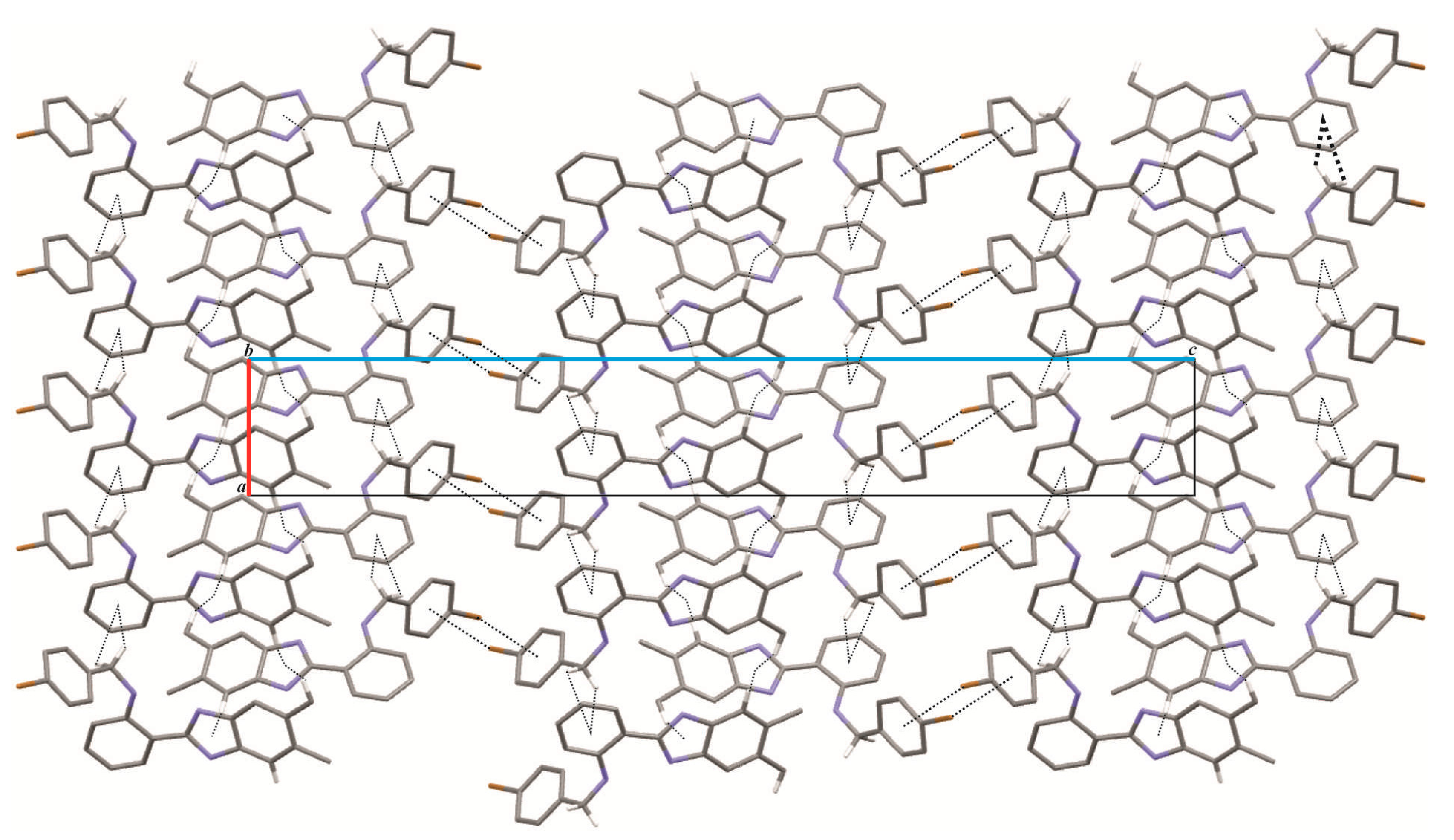
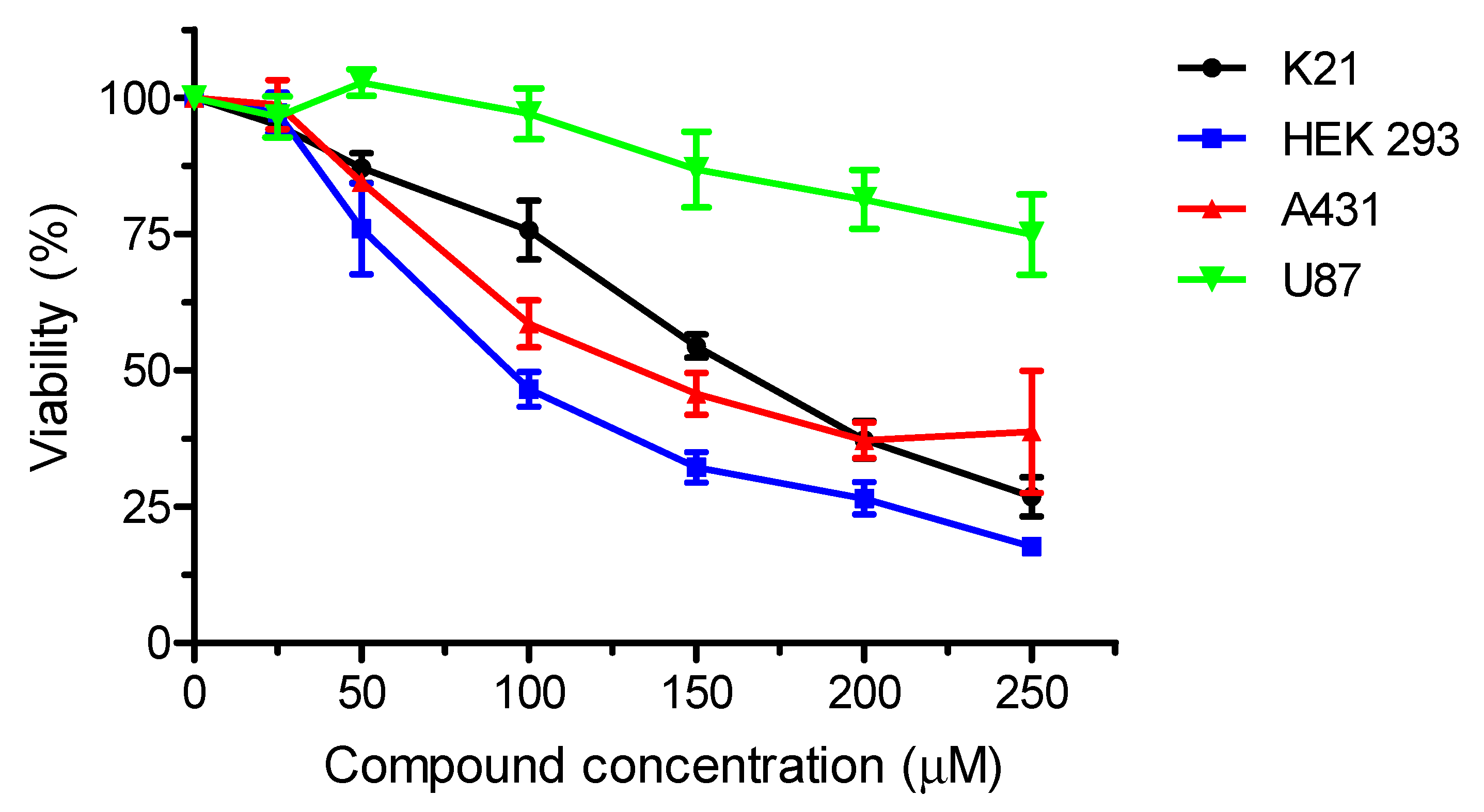
2. Results and Discussion
3. Materials and Methods
Synthesis of N-(4-Bromobenzyl)-2-(5,6-dimethyl-1H-benzo[d]imidazol-2-yl)benzeneamine (15)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Salahuddin, M.; Shaharyar, M.; Mazumder, A. Benzimidazoles: A biologically active compounds. Arab. J. Chem. 2017, 10, S157–S173. [Google Scholar] [CrossRef]
- Keri, R.S.; Rajappa, C.K.; Patil, S.A.; Nagaraja, B.M. Benimidazole-core as an antimycobacterial agent. Pharmacol. Rep. 2016, 68, 1254–1265. [Google Scholar] [CrossRef] [PubMed]
- Bansal, Y.; Silakari, O. The therapeutic journey of benzimidazoles: A review. Bioorg. Med. Chem. 2012, 20, 6208–6236. [Google Scholar] [CrossRef] [PubMed]
- Alaqeel, S.I. Synthetic approaches to benzimidazoles from o-phenylenediamine: A literature review. J. Saudi Chem. Soc. 2017, 21, 229–327. [Google Scholar] [CrossRef]
- Keri, R.S.; Hiranathad, A.; Budagumpi, S.; Nagaraja, B.M. Comprehensive Review in Current Developments of Benzimidazole—Based Medicinal Chemistry. Chem. Biol. Drug Des. 2015, 86, 19–65. [Google Scholar] [CrossRef] [PubMed]
- Yadav, G.; Ganguly, S. Structure activity relationship (SAR) study of benzimidazole scaffold for different biological activities: A mini-review. Eur. J. Med. Chem. 2015, 97, 419–443. [Google Scholar] [CrossRef] [PubMed]
- Ajani, O.O.; Aderohunmu, D.V.; Ikpo, C.O.; Adedapo, A.E.; Olanrewaju, I.O. Functionalized Benzimidazole Scaffolds: Privileged Heterocycle for Drug Design in Therapeutic Medicine. Arch. Pharm. Chem. Life Sci. 2016, 349, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Barot, K.P.; Mikolova, S.; Ivanov, I.; Ghate, M.D. Novel research strategies of benzimidazole derivatives: A review. Mini-Rev. Med. Chem. 2013, 13, 1421–1447. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Han, X.; Zhou, Z. New substituted benzimidazole derivatives: A patent review (2013–2014). Expert Opin. Ther. Pat. 2015, 25, 595–612. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Fei, F. New substituted benzimidazole derivatives: A patent review (2010–2012). Expert Opin. Ther. Pat. 2013, 23, 1157–1179. [Google Scholar] [CrossRef]
- Coppola, G.M. The chemistry of isatoic anhydride. Synthesis 1980, 23, 505–536. [Google Scholar] [CrossRef]
- Kappe, T.; Stadlbauer, W. Isatoic anhydrides and their uses in heterocyclic synthesis. Adv. Heterocycl. Chem. 1981, 28, 127–182. [Google Scholar] [CrossRef]
- Shvekhgeimer, M.-G.A. Synthesis of heterocyclic compounds based on isatoic anhydrides (2H-3,1-benzoxazine-2,4-diones). (Review). Chem. Heterocycl. Compd. 2001, 37, 385–443. [Google Scholar] [CrossRef]
- Tylor, E.C.; Yoneda, F. Condensed imidazoles from o-diamines and isatoic anhydrides. Angew. Chem. Int. Ed. 1967, 6, 878–879. [Google Scholar] [CrossRef]
- Padmaja, J.; Reddy, M.S.; Ratnam, C.V. Reaction of 1,2-dihydronaph[1,2-d]oxazin-2,4(H)-dione with ortho-substituted anilines. Ind. J. Chem. 1987, 26, 951–954. [Google Scholar]
- Devi, K.R.; Reddy, M.S. Synthesis of 6,7-dihydro-6-substituted benzimidazo[1,2-c]benzo[g]quinazolines and their heteroaromatic analogues. Ind. J. Chem. 1994, 33b, 1013–1016. [Google Scholar] [CrossRef]
- Bahekar, R.H.; Ram Rao, A.R. New broncholidators—Synthesis of 6-alkylbenzimidazo[1,2-c]quinazolines. Indian J. Pharm. Sci. 2000, 62, 41–45. [Google Scholar]
- Fadda, A.A.; Refat, H.M.; Zaki, M.E.A.; Monir, E. Reaction of isatoic anhydride with bifunctional reagents: Synthesis of Some new quinazolone fused heterocycles, 2-substituted anilinoheterocyclic derivatives and other related compounds. Synth. Commun. 2001, 31, 3537–3545. [Google Scholar] [CrossRef]
- Yoneda, F.; Mera, F. Reaction of 5,6-diamino-1,3-dimethyluracil with isatoic anhydrides. Chem. Pharm. Bull. 1972, 20, 1815–1818. [Google Scholar] [CrossRef]
- Mieczkowski, A.; Bazlekova, M.; Bagiński, M.; Wójcik, J.; Winczura, A.; Miazga, A.; Gajda, R.; Woźniak, K.; Tudek, B. A mild and efficient approach to 6H-oxazolo[3,2-f]pyrimidine-5,7-dione scaffold via unexpected rearrangement of 2,3-dihydropyrimido[6,1-b][1,5,3]dioxazepine-7,9(5H,8H)-diones: A synthesis, crystallographic studies and cytotoxic activity screening. Tetrahedron Lett. 2016, 57, 743–746. [Google Scholar] [CrossRef]
- Mieczkowski, A.; Trzybiński, D.; Wilczek, M.; Psurski, M.; Bagiński, M.; Bieszczad, B.; Mroczkowska, M.; Woźniak, K. (S)-2-(4-Chlorobenzoyl)-1,2,3,4-tetrahydrobenzo[e]pyrazino[1,2-a][1,4]diazepine-6,12(11H,12aH)-dione—Synthesis and Crystallographic Studies. Molbank 2017, 2017, M964. [Google Scholar] [CrossRef]
- Mieczkowski, A.; Wińska, P.; Kaczmarek, M.; Mroczkowska, M.; Garbicz, D.; Pilżys, T.; Marcinkowski, M.; Piwowarski, J.; Grzesiuk, E. 2′-Deoxy-2′-azidonucleoside analogues: Synthesis and evaluation of antitumor and antimicrobial activity. Chem. Pap. 2017. [Google Scholar] [CrossRef]
- Hardtmann, G.E.; Koleta, G.; Pfister, O.R. Chemistry of 2H-3,1-benzoxazine-2,4(1H)-dione (isatoic anhydrides) 1. Synthesis of N-substituted 2H-3,1-benzoxazine-2,4(1H)-diones. J. Heterocycl. Chem. 1975, 12, 565–572. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- CrysAlis CCD. CrysAlis RED; Oxford Diffraction Ltd.: Yarnton, UK, 2008. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidock, E.; Rodriguez-Monge, L.; Tylor, J.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dziełak, M.; Trzybiński, D.; Czerwińska, J.; Majchrzak, B.; Tudek, B.; Woźniak, K.; Mieczkowski, A. N-(4-Bromobenzyl)-2-(5,6-dimethyl-1H-benzo[d]imid-azol-2-yl)benzeneamine. Molbank 2018, 2018, M979. https://doi.org/10.3390/M979
Dziełak M, Trzybiński D, Czerwińska J, Majchrzak B, Tudek B, Woźniak K, Mieczkowski A. N-(4-Bromobenzyl)-2-(5,6-dimethyl-1H-benzo[d]imid-azol-2-yl)benzeneamine. Molbank. 2018; 2018(1):M979. https://doi.org/10.3390/M979
Chicago/Turabian StyleDziełak, Monika, Damian Trzybiński, Jolanta Czerwińska, Bartosz Majchrzak, Barbara Tudek, Krzysztof Woźniak, and Adam Mieczkowski. 2018. "N-(4-Bromobenzyl)-2-(5,6-dimethyl-1H-benzo[d]imid-azol-2-yl)benzeneamine" Molbank 2018, no. 1: M979. https://doi.org/10.3390/M979
APA StyleDziełak, M., Trzybiński, D., Czerwińska, J., Majchrzak, B., Tudek, B., Woźniak, K., & Mieczkowski, A. (2018). N-(4-Bromobenzyl)-2-(5,6-dimethyl-1H-benzo[d]imid-azol-2-yl)benzeneamine. Molbank, 2018(1), M979. https://doi.org/10.3390/M979