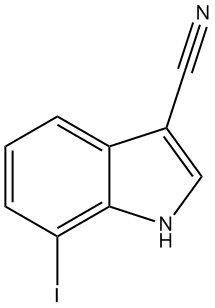
7-Iodo-1H-indole-3-carbonitrile


Abstract
:
{kind=link}
{kind=link}
1. Introduction
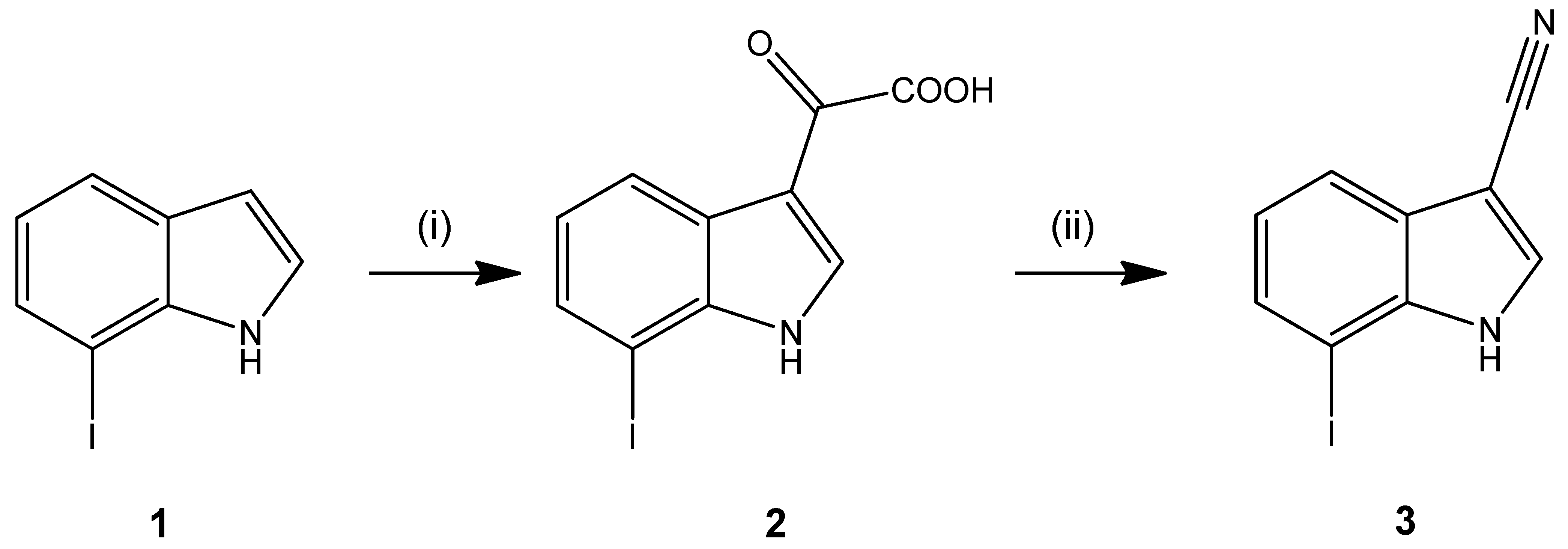
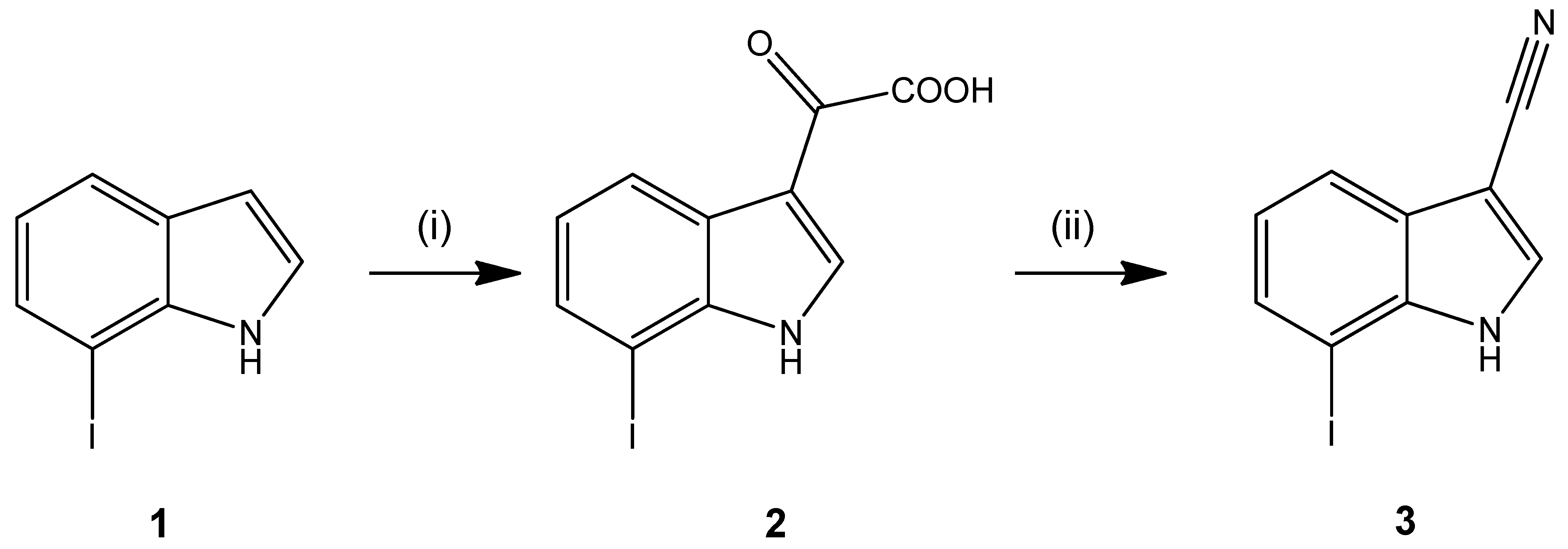
2. Results and Discussion
3. Experimental
3.1. General
3.2. 7-Iodo-1H-indole-3-carbonitrile (3)
Supplementary materials
Supplementary File 1Supplementary File 2Supplementary File 3Supplementary File 4Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References and Notes
- Fabbro, D. 25 years of small molecular weight kinase inhibitors: potentials and limitations. Mol. Pharmacol. 2015, 87, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Kunick, C.; Egert-Schmidt, A. Die kurze Geschichte der Proteinkinase-Inhibitoren. Jung, kompetitiv, erfolgreich. Pharmazie in Unserer Zeit 2008, 37, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol. Res. 2015, 100, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Rask-Andersen, M.; Zhang, J.; Fabbro, D.; Schioth, H.B. Advances in kinase targeting: Current clinical use and clinical trials. Trends Pharmacol. Sci. 2014, 35, 604–620. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, L.A.; Todd, D.J. Kinase inhibitors: The next generation of therapies in the treatment of rheumatoid arthritis. Int. J. Rheum. Dis. 2014, 17, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Gadina, M. Advances in kinase inhibition: Treating rheumatic diseases and beyond. Curr. Opin. Rheumatol. 2014, 26, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Abbassi, R.; Johns, T.G.; Kassiou, M.; Munoz, L. DYRK1A in neurodegeneration and cancer: Molecular basis and clinical implications. Pharmacol. Ther. 2015, 151, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Karthikeyan, C.; Moorthy, N.S.; Waiker, D.K.; Jain, A.K.; Trivedi, P. Human CDC2-like kinase 1 (CLK1): A novel target for Alzheimer's disease. Curr. Drug Targets 2014, 15, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Egert-Schmidt, A.-M.; Dreher, J.; Dunkel, U.; Kohfeld, S.; Preu, L.; Weber, H.; Ehlert, J.E.; Mutschler, B.; Totzke, F.; Schächtele, C.; et al. Identification of 2-anilino-9-methoxy-5,7-dihydro-6H-pyrimido[5,4-d][1]benzazepin-6-ones as dual PLK1/VEGF-R2 kinase inhibitor chemotypes by structure-based lead generation. J. Med. Chem. 2010, 53, 2433–2442. [Google Scholar] [CrossRef] [PubMed]
- Falke, H.; Bumiller, K.; Harbig, S.; Masch, A.; Wobbe, J.; Kunick, C. 2-tert-Butyl-5,6,7,8,9,10-hexahydrocyclohepta[b]indole. Molbank 2011, 2011, M737. [Google Scholar] [CrossRef]
- McGrath, C.F.; Pattabiraman, N.; Kellogg, G.E.; Lemcke, T.; Kunick, C.; Sausville, E.A.; Zaharevitz, D.W.; Gussio, R. Homology model of the CDK1/cyclin B complex. J. Biomol. Struct. Dyn. 2005, 22, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Orban, O.C.F.; Korn, R.S.; Unger, L.; Yildiz, A.; Kunick, C. 3-Chlorokenpaullone. Molbank 2015, 2015, M856. [Google Scholar] [CrossRef]
- Tolle, N.; Kunick, C. Paullones as inhibitors of protein kinases. Curr. Top. Med. Chem. 2011, 11, 1320–1332. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Preu, L.; Lemcke, T.; Totzke, F.; Schächtele, C.; Kubbutat, M.H.G.; Kunick, C. Dual IGF-1R/SRC inhibitors based on a N′-aroyl-2-(1H-indol-3-yl)-2-oxoacetohydrazide structure. Eur. J. Med. Chem. 2011, 46, 2759–2769. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, O.; Huber, K.; Eisenreich, A.; Filippakopoulos, P.; King, O.; Bullock, A.N.; Szklarczyk, D.; Jensen, L.J.; Fabbro, D.; Trappe, J.; et al. Specific CLK inhibitors from a novel chemotype for regulation of alternative splicing. Chem. Biol. 2011, 18, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Poznański, J.; Shugar, D. Halogen bonding at the ATP binding site of protein kinases: Preferred geometry and topology of ligand binding. Biochim. Biophys. Acta 2013, 1834, 1381–1386. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wasik, R.; Winska, P.; Poznanski, J.; Shugar, D. Synthesis and physico-chemical properties in aqueous medium of all possible isomeric bromo analogues of benzo-1H-triazole, potential inhibitors of protein kinases. J. Phys. Chem. B 2012, 116, 7259–7268. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, Y.; Wang, J. Lewis acid catalyzed direct cyanation of indoles and pyrroles with N-cyano-N-phenyl-p-toluenesulfonamide (NCTS). Org. Lett. 2011, 13, 5608–5611. [Google Scholar] [CrossRef] [PubMed]
- Borsche, W. Zur Kenntnis der Benzisoxazole. Liebigs Ann. Chem. 1912, 390, 1–29. [Google Scholar] [CrossRef]
- Characterization of 2: M.p.: 222–223 °C (dec.); MS (EI, rel intensity) m/z (%): 315 ([M]+•, 27), 270 (100), 242 (5); IR (KBr) (cm−1): 3241 (NH/OH), 1745, 1617 (C=O); 1H-NMR (400.4 MHz, DMSO-d6): δ (ppm) = 7.08 (t, 1H, J = 7.8 Hz, C(5)H), 7.7 (dd, 1H, J = 7.5/1.0 Hz, C(6)H), 8.20 (dt, 1H, J = 7.6/0.6 Hz, C(4)H), 8.37 (d, 1H, J = 3.4 Hz, C(2)H), 12.30 (d, 1H, J = 3.5 Hz, NH-indole), 14.00 (s, 1H, COOH); 13C-NMR (100.7 MHz, DMSO-d6): δ (ppm) = 121.1, 124.5, 132.7, 138.1 (CH), 77.96 (C(7)), 113.2, 126.2, 138.6 (C), 164.7 (COOH), 180.8 (C=O); HPLC (AUC%): 99.42% at 254 nm; 99.66% at 280 nm; tms = 2.97 min, ts (DMSO) = 1.16 min (isocratic); TLC (toluene/ethyl acetate/formic acid 10:1:1): Rf = 0.26; Anal. calculated for C10H6INO3 (315.07): C, 38.12; H, 1.92; N, 4.45. Found: C, 37.93; H, 1.80; N, 4.42.
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meine, R.; Falke, H.; Kötz, J.; Schweda, S.I.; Kunick, C. 7-Iodo-1H-indole-3-carbonitrile. Molbank 2015, 2015, M869. https://doi.org/10.3390/M869
Meine R, Falke H, Kötz J, Schweda SI, Kunick C. 7-Iodo-1H-indole-3-carbonitrile. Molbank. 2015; 2015(4):M869. https://doi.org/10.3390/M869
Chicago/Turabian StyleMeine, Rosanna, Hannes Falke, Jana Kötz, Sandra I. Schweda, and Conrad Kunick. 2015. "7-Iodo-1H-indole-3-carbonitrile" Molbank 2015, no. 4: M869. https://doi.org/10.3390/M869
APA StyleMeine, R., Falke, H., Kötz, J., Schweda, S. I., & Kunick, C. (2015). 7-Iodo-1H-indole-3-carbonitrile. Molbank, 2015(4), M869. https://doi.org/10.3390/M869