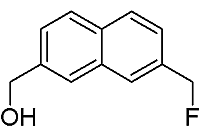
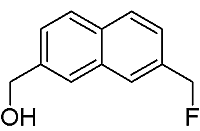
[7-(Fluoromethyl)-2-naphthyl]methanol

Abstract
:
{kind=link}
{kind=link}
Introduction
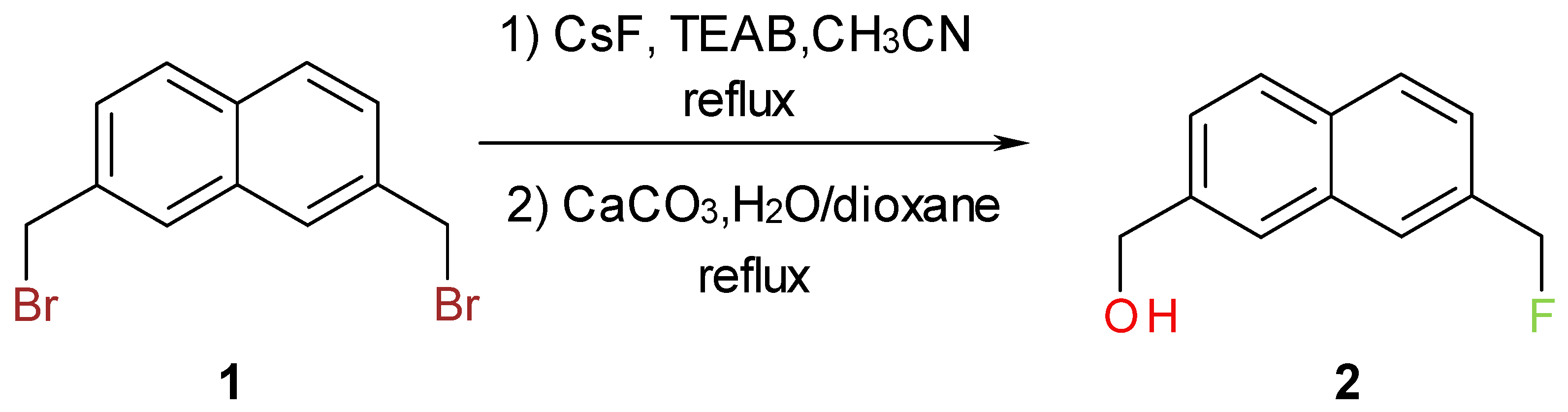
Results and Discussion
Experimental Section
General
Synthesis of [7-(Fluoromethyl)-2-naphthyl]methanol
Supplementary materials
Supplementary File 1Supplementary File 2Supplementary File 3Acknowledgments
Conflicts of Interest
References and Notes
- Filler, R. Organofluorine compounds in medicinal chemistry and biomedical applications. In Studies in Organic Chemistry; Filler, R., Ed.; Elsevier: New York, NY, USA, 1993; Volume 48, pp. 1–23. [Google Scholar]
- Seebach, D. Organic synthesis-Where now? Angew. Chem. Int. Ed. Engl. 1990, 29, 1320–1367. [Google Scholar] [CrossRef]
- Yamamoto, H.; Hiyama, T. Biologically active organofluorine compounds. In Organofluorine Compounds; Springer: Berlin, Germany; Heidelberg, Germany, 2000; pp. 137–182. [Google Scholar]
- Olah, G.A.; Nojima, M.; Kerekes, I. Synthetic methods and reactions IX. Fluorination of secondary- and tertiary-alcohols with polyhydrogen fluoride/pyridine(trialkylamine) reagents. Synthesis 1973, 786–787. [Google Scholar] [CrossRef]
- Minami, A.; Uchida, R.; Eguchi, T.; Kakinuma, K. Enzymatic approach to unnatural glycosides with diverse aglycon scaffolds using glycosyltransferase VinC. J. Am. Chem. Soc. 2005, 127, 6148–6149. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.H. Fluoride ion as a base in organic synthesis. Chem. Rev. 1980, 80, 429–452. [Google Scholar] [CrossRef]
- Gerstenberger, M.R.C.; Haas, A. Methods of fluorination in organic chemistry. Angew. Chem. Int. Ed. Engl. 1981, 20, 647–667. [Google Scholar] [CrossRef]
- Mascaretti, O.A. Modern methods for the monofluorination of aliphatic organic compounds. Aldrichim. Acta 1993, 26, 47–58. [Google Scholar]
- Bhadury, P.S.; Pandey, M.; Jaiswal, D.K. A facile synthesis of organofluorine compounds using a semi-molten mixture of tetrabutylammonium bromide and an alkali metal fluoride. J. Fluorine Chem. 1995, 73, 185–187. [Google Scholar] [CrossRef]
- Gingras, M.; N. Harpp, D. New anhydrous fluorinating systems: The combination of crown-ethers and cesium fluoride. A relative rate study. Tetrahedron Lett. 1988, 29, 4669–4672. [Google Scholar] [CrossRef]
- Kim, D.W.; Song, C.E.; Chi, D.Y. Significantly enhanced reactivities of the nucleophilic substitution reactions in ionic liquid. J. Org. Chem. 2003, 68, 4281–4285. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.K.; Jeong, H.J.; Seok, T.L.; Sohn, M.H.; Katzenellenbogen, J.A.; Dae, Y.C. Facile nucleophilic fluorination reactions using tert-alcohols as a reaction medium: Significantly enhanced reactivity of alkali metal fluorides and improved selectivity. J. Org. Chem. 2008, 73, 957–962. [Google Scholar]
- Shinde, S.S.; Lee, B.S.; Chi, D.Y. Synergistic effect of two solvents, tert-alcohol and ionic liquid, in one molecule in nucleophilic fluorination. Org. Lett. 2008, 10, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Dixon, E.A.; Fischer, A.; Robinson, F.P. Preparation of a series of substituted fluoromethyl-naphthalenes. Can. J. Chem. 1981, 59, 2629–2641. [Google Scholar] [CrossRef]
- Adcock, W.; Abeywickrema, A.N. Conformational preference of the fluoromethyl group in some benzyl fluorides: A 13C N.M.R. study. Aust. J. Chem. 1980, 33, 181–187. [Google Scholar] [CrossRef]
- Still, W.C.; Kahn, M.; Mitra, A. Rapid chromatographic technique for preparative separations with moderate resolution. J. Org. Chem. 1978, 43, 2923–2925. [Google Scholar] [CrossRef]
- Ried, W.; Bodem, H. Characteristic derivatives of the di- and trimethylnaphthalenes. Chem. Ber. 1958, 91, 1981–1982. [Google Scholar] [CrossRef]
© 2014 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bogdanov, J. [7-(Fluoromethyl)-2-naphthyl]methanol. Molbank 2014, 2014, M818. https://doi.org/10.3390/M818
Bogdanov J. [7-(Fluoromethyl)-2-naphthyl]methanol. Molbank. 2014; 2014(1):M818. https://doi.org/10.3390/M818
Chicago/Turabian StyleBogdanov, Jane. 2014. "[7-(Fluoromethyl)-2-naphthyl]methanol" Molbank 2014, no. 1: M818. https://doi.org/10.3390/M818
APA StyleBogdanov, J. (2014). [7-(Fluoromethyl)-2-naphthyl]methanol. Molbank, 2014(1), M818. https://doi.org/10.3390/M818