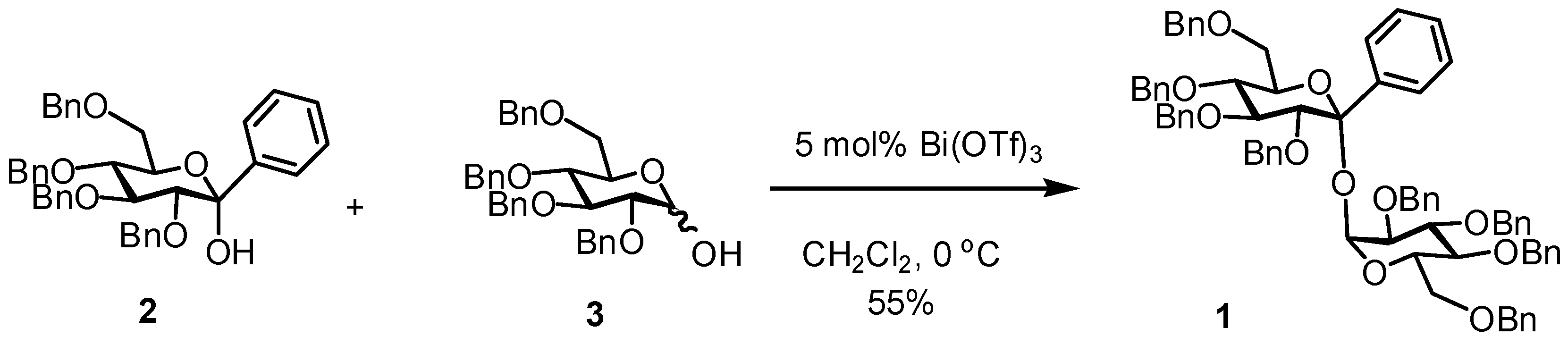
Abstract
The title compound 1 was synthesized by the coupling reaction of 2,3,4,6-tetra-O-benzyl-1-C-phenyl-α-d-glucopyranose (2) with 2,3,4,6-tetra-O-benzyl-d-glucopyranose (3) in the presence of 5 mol% bismuth(III) triflate in dichloromethane at 0 °C.
Trehalose is a non-reducing disaccharide which is composed of two glucopyranosyl units linked to each other in α-configuration. Trehalose is well-known for its various biological functions, and current attention is focused on the biologically novel functions of trehalose analogs, such as binding potentials to E-selectin and shiga toxins, and antibacterial activities [1,2,3]. Therefore, it is of increasing importance to design and synthesize novel non-reducing disaccharides which are structurally-classified as trehalose analogs.
Our recent studies showed the synthetic approaches to two types of non-reducing disaccharides from aldoses and/or ketoses by bismuth(III) triflate (Bi(OTf)3)-catalyzed dehydrative glycosidations [4,5,6]. One was non-reducing disaccharides composed of two aldoses which linked with each other by self-condensations [5]. The other was a hybrid type of non-reducing disaccharides composed of aldose and ketose by the cross-condensations [6]. The latter condensation was the ketosylation reaction that utilized the ketohexopyranoses carrying the methyl group at the anomeric centers, i.e., the 1-C-methyl-hexopyranose derivatives, as the glycosyl donors by using only 5 mol% Bi(OTf)3. Since some ketohexopyranose derivatives having a functional group such as an alkyl, alkynyl, alkene, and aryl group at the anomeric carbon centers are readily prepared from the corresponding glycono-1,5-lactone derivatives and appropriate organometallic reagents, it is expected that various kinds of structurally novel non-reducing disaccharides can be created using such ketohexopyranose derivatives. We also achieved the synthesis of a non-reducing disaccharide using the 1-C-butyl-d-glucopyranose derivative employing the same procedure [7].
We designed a novel non-reducing disaccharide using the 1-C-phenyl-d-glucopyranose derivative as a trehalose analog. This paper describes the synthesis of 2,3,4,6-tetra-O-benzyl-1-C-phenyl-d-glucopyranosyl 2,3,4,6-tetra-O-benzyl-d-glucopyranoside (1) by the Bi(OTf)3-catalyzed ketosylation of 2,3,4,6-tetra-O-benzyl-1-C-phenyl-d-glucopyranose (2) [8,9] into 2,3,4,6-tetra-O-benzyl-d-glucopyranose (3) [10].
The ketosylation of 2 with 3 was investigated using Bi(OTf)3 as shown in Scheme 1. The condensation reaction of 2 with 3 in the presence of 5 mol% Bi(OTf)3 in dichloromethane at 0 °C for 15 h afforded 1 in a good yield of 55%. The benzyl 2,3,4,6-tetra-O-benzyl-1-C-phenyl-α-d-glucopyranoside (4) was obtained in 11% yield as a major by-product. Compound 4 would be formed by the reaction of 2 with benzyl alcohol generated by the degradation of 2 and/or 3. A similar reaction at room temperature reduced the yield of 1 (12%) and increased the yield of 4 (26%).
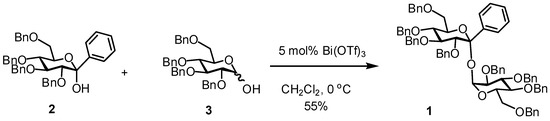
Scheme 1.
Synthesis of the title compound 1.
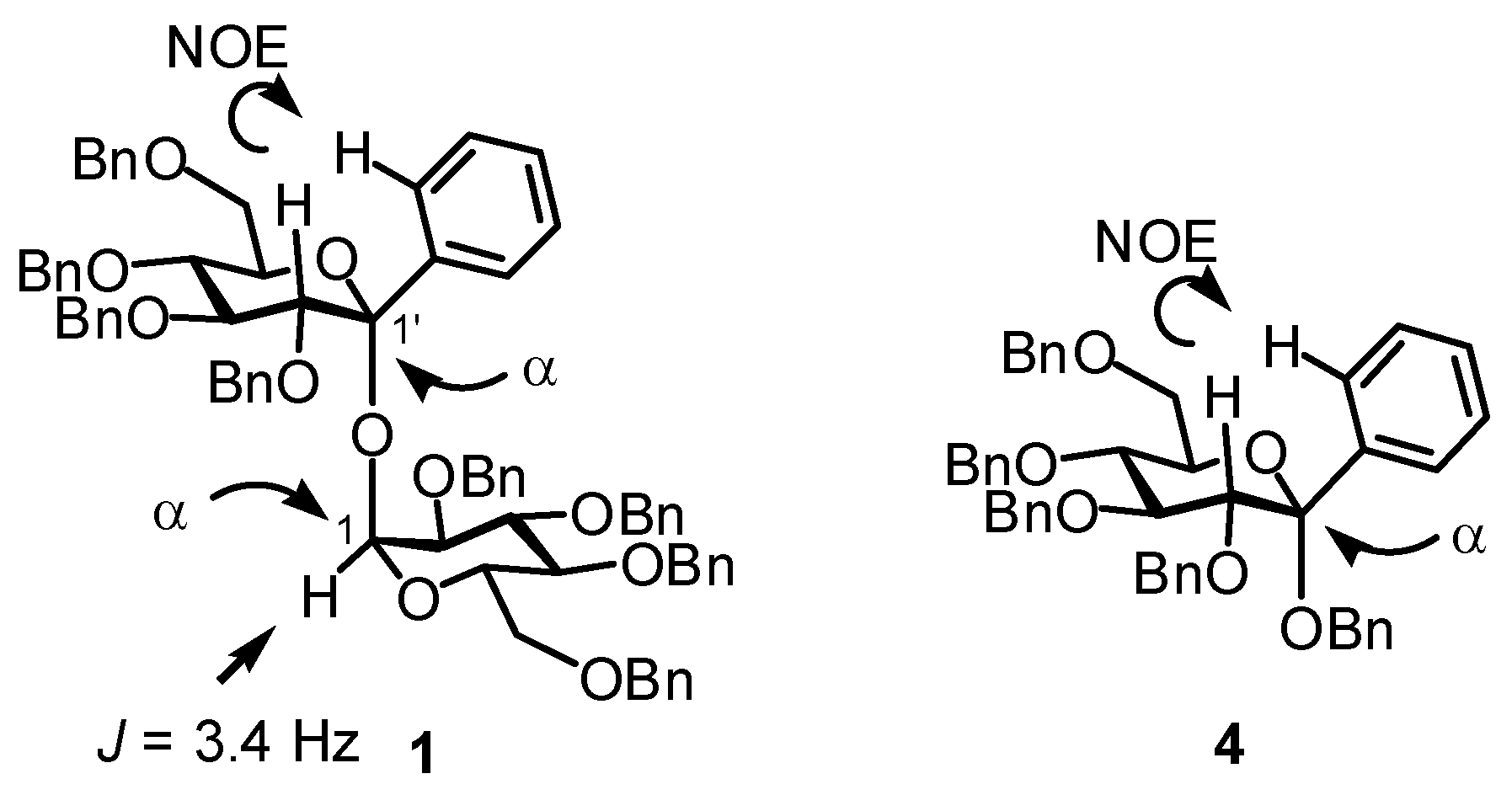
The configuration of the two glycosidic linkages of 1 was determined by NOE and 1H-NMR experiments. The NOE interaction between aromatic-ortho H and H-2' as shown in Figure 1 was observed. This observation inevitably indicated the equatorial orientation of the phenyl group and determined the α-ketopyranosidic linkage. The α-aldopyranosidic linkage was confirmed by the coupling constant (J = 3.4 Hz) of H-1. Thus, both the glycosidic linkages of 1 were α. The ketopyranosidic linkage of 4 was similarly determined as an α by the NOE interaction.
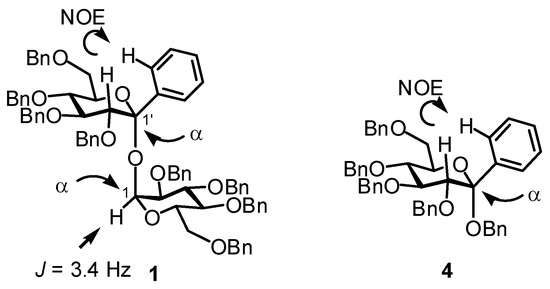
Figure 1.
NOE interaction.
Experimental
2,3,4,6-Tetra-O-benzyl-1-C-phenyl-α-d-glucopyranosyl 2,3,4,6-tetra-O-benzyl-α-d-glucopyranoside (1)
To a suspension of Bi(OTf)3 (4.7 mg, 0.007 mmol), 2 (47.3 mg, 0.09 mmol) and CaSO4 (ca. 100 mg) in CH2Cl2 (3.5 mL) was added 3 (83.2 mg, 0.13 mmol) at 0 °C under an Ar atmosphere. The resulting mixture was stirred for 15 h. The reaction was then quenched by addition of a sat. NaHCO3 solution (5 mL). The reaction mixture was extracted with CH2Cl2, and the organic layer was washed with water and a sat. NaCl solution. After the organic layer was dried over Na2SO4, the solvent was evaporated under reduced pressure. The crude product was purified by preparative silica gel TLC (ethyl acetate/hexane = 1/3) to give 1 (Rf = 0.58, 55.3 mg, 55%) and 4 (Rf = 0.65, 10.1 mg, 11%) both as colorless oils. Compound 1: [α]D21 +49° (c 0.74, CHCl3). 1H-NMR (600 MHz, CDCl3): δ 3.32 (1H, d, J = 9.7 Hz, H-2'), 3.33–3.37 (2H, m, Ha-6, Ha-6'), 3.42 (1H, dd, J = 3.4 Hz, J = 10.3 Hz, H-2), 3.47 (1H, dd, J = 2.8 Hz, J = 11.0 Hz, Hb-6'), 3.62–3.64 (1H, m, Hb-6), 3.65 (1H, t, J = 8.9 Hz, H-4), 3.80 (1H, t, J = 9.6 Hz, H-4'), 4.121 (1H, t, J = 9.6 Hz, H-3), 4.124 (1H, d, J = 11.0 Hz, CH2Ph), 4.16 (1H, t, J = 9.7 Hz, H-3'), 4.28–4.30 (1H, m, H-5), 4.35 (1H, d, J = 12.3 Hz, CH2Ph), 4.44–4.62 (8H, m, H-5', CH2Ph), 4.75 (1H, d, J = 11.0 Hz, CH2Ph), 4.83–4.92 (5H, m, CH2Ph), 4.99 (1H, d, J = 11.0 Hz, CH2Ph), 5.03 (1H, d, J = 3.4 Hz, H-1), 7.14–7.33 (43H, m, Ph), 7.83 (2H, d, J = 7.6 Hz, Armatic ortho-H). 13C-NMR (150 MHz, CDCl3): δ 68.2 (C-6), 68.6 (C-6'), 70.5 (C-5), 71.4 (C-5'), 73.2 (CH2Ph), 73.3 (CH2Ph), 73.4 (CH2Ph), 74.7 (CH2Ph), 75.0 (CH2Ph), 75.1 (CH2Ph), 75.47 (CH2Ph), 75.51 (CH2Ph), 78.21 (C-4), 78.22 (C-4'), 80.5 (C-2), 81.8 (C-3), 83.1 (C-3'), 85.5 (C-2'), 92.1 (C-1), 101.3 (C-1'), 127.1–128.4 (Ph), 138.0–138.7 (Ph). HRMS (ESI): m/z calcd for C74H74O11Na+: 1161.5123; found: 1161.5142. Anal. Calcd for C74H74O11·H2O: C, 76.79; H, 6.62. Found: C, 76.97; H, 6.82. Compound 4: [α]D24 +41° (c 0.55, CHCl3). 1H-NMR (600 MHz, CDCl3): δ 3.38 (1H, d, J = 9.6 Hz, H-2), 3.74 (1H, d, J = 10.3 Hz, Ha-6), 3.80–3.85 (3H, m, H-4, Hb-6, CH2Ph), 3.90 (1H, dd, J = 3.4 Hz, J = 10.3 Hz, H-5), 4.25 (1H, t, J = 8.9 Hz, H-3), 4.37 (2H, s, CH2Ph), 4.46 (1H, d, J = 10.3 Hz, CH2Ph), 4.61 (1H, d, J = 12.3 Hz, CH2Ph), 4.63 (1H, d, J = 13.1 Hz, CH2Ph), 4.70 (1H, d, J = 12.4 Hz, CH2Ph), 4.86–4.88 (2H, m, CH2Ph), 4.94 (1H, d, J = 11.0 Hz, CH2Ph), 7.10–7.41 (28H, m, Ph), 7.72 (2H, d, J = 8.2 Hz, Armatic ortho-H). 13C-NMR (150 MHz, CDCl3): δ 63.7 (CH2Ph), 68.9 (C-6), 72.3 (C-5), 73.4 (CH2Ph), 75.1 (CH2Ph), 75.5 (CH2Ph), 75.7 (CH2Ph), 78.6 (C-4), 83.1 (C-3), 85.8 (C-2), 101.5 (C-1), 127.3–128.4 (Ph), 138.3–138.8 (Ph). HRMS (ESI): m/z calcd for C47H46O6Na+: 729.3187; found: 729.3216. Anal. Calcd for C47H46O6·2/3H2O: C, 78.53; H, 6.64. Found: C, 78.42; H, 6.61.
Supplementary materials
Supplementary File 1Supplementary File 2Supplementary File 3References and Notes
- Hiruma, K.; Kajimoto, T.; Weitz-Schmidt, G.; Ollmann, I.; Wong, C.-H. Rational design and synthesis of a 1,1-linked disaccharide that is 5 times as active as sialyl Lewis X in binding to E-selectin. J. Am. Chem. Soc. 1996, 118, 9265–9270. [Google Scholar] [CrossRef]
- Dohi, H.; Nishida, Y.; Furuta, Y.; Uzawa, H.; Yokoyama, S.-I.; Ito, S.; Mori, H.; Kobayashi, K. Molecular design and biological potential of galacto-type trehalose as a nonnatural ligand of shiga toxins. Org. Lett. 2002, 4, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Hui, Y.; Chang, C.-H.T. Convenient divergent synthesis of a library of trehalosamine analogues. Org. Lett. 2002, 4, 2245–2248. [Google Scholar] [CrossRef] [PubMed]
- Yamanoi, T.; Inoue, R.; Matsuda, S.; Hamasaki, K. Bismuth(III) triflate-catalyzed dehydrative glycosidation using 1-hydroxy sugars. Lett. Org. Chem. 2008, 5, 30–33. [Google Scholar] [CrossRef]
- Yamanoi, T.; Inoue, R.; Matsuda, S.; Iwao, K.; Oda, Y.; Yoshida, A.; Hamasaki, K. Formation of O-glycosidic linkages from 1-hydroxy sugars by bismuth(III) triflate-catalyzed dehydrative glycosidation. Heterocycles 2009, 77, 445–460. [Google Scholar] [CrossRef]
- Yamanoi, T.; Inoue, R.; Matsuda, S.; Katsuraya, K.; Hamasaki, K. Synthesis of trehalose mimics by bismuth(III) triflate or bis(trifluoromethane)sulfonimide-catalyzed 1-C-methyl-d-hexopyranosylation. Tetrahedron Asymmetry 2006, 17, 2914–2918. [Google Scholar] [CrossRef]
- Yamanoi, T.; Inoue, R.; Oda, Y.; Hamasaki, K. 6,7,8,10-Tetra-O-benzyl-1,2,3,4-tetradeoxy-α-d-gluco-dec-5-ulopyranosyl 2,3,4,6-tetra-O-benzyl-α-d-glucopyranoside. Molbank 2010, 2010, M671. [Google Scholar] [CrossRef]
- Ellsworth, B.-A.; Doyle, A.-G.; Patel, M.; Caceres-Cortes, J.; Meng, W.; Deshpande, P.-P.; Pullockaran, A.; Washburn, W.-N. C-Arylglucoside synthesis: Triisopropylsilane as a selective reagent for the reduction of an anomeric C-phenyl ketal. Tetrahedron Asymmetry 2003, 14, 3243–3247. [Google Scholar] [CrossRef]
- Czernecki, S.; Ville, G. C-Glycosides. 7. Stereospecific C-glycosylation of aromatic and heterocyclic rings. J. Org. Chem. 1989, 54, 610–612. [Google Scholar] [CrossRef]
- Yamanoi, T.; Misawa, N.; Matsuda, S.; Watanabe, M. Preparation of partially benzylated mono-, di-, and trisaccharides by selective cleavage of the β-fructofuranosidic linkage in fully benzylated sucrose and sucrose-related oligosaccharides under acidic conditions. Carbohydr. Res. 2008, 343, 1366–1372. [Google Scholar] [CrossRef] [PubMed]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).