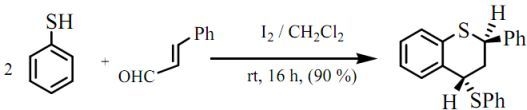
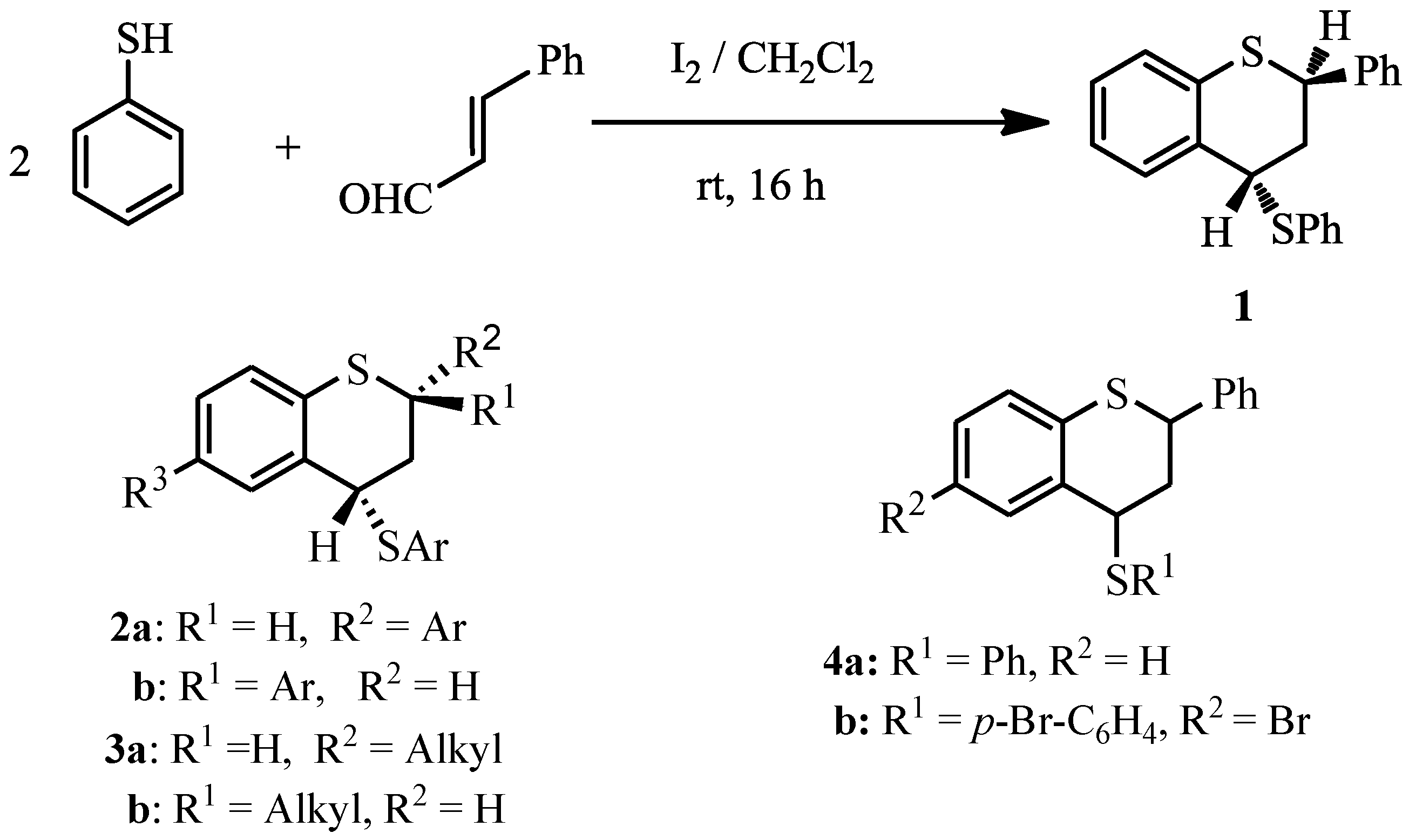
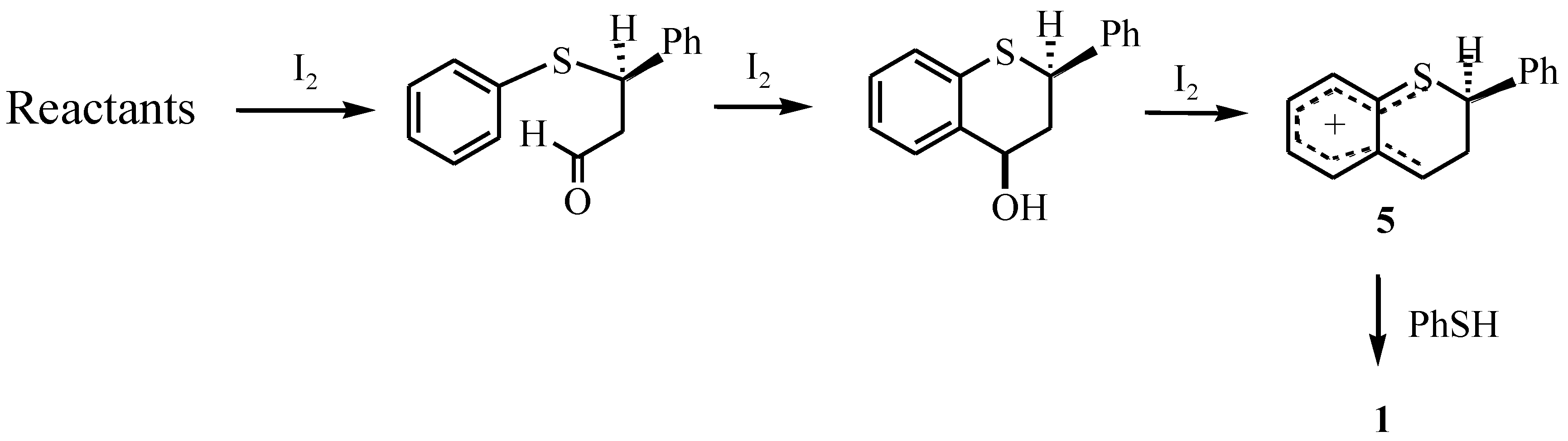
trans-2-Phenyl-4-thiophenoxy-3,4-dihydro-2H-1-benzothiopyran
Abstract
:
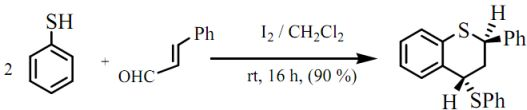{kind=link}
{kind=link}
{kind=link}
{kind=link}
Introduction
Results and Discussion

Experimental Section
Conclusions
Supplementary materials
Supplementary File 1Supplementary File 2Supplementary File 3Acknowledgements
References and Notes
- Wang, W.; Li, H.; Wang, J.; Zu, L. Enantioselective organocatalytic tandem Michael-aldol reactions: One-pot synthesis of chiral thiochromenes. J. Amer. Chem. Soc. 2006, 128, 10354–10355, and references cited therein. [Google Scholar] [CrossRef] [PubMed]
- Ishino, Y.; Mihara, M.; Kawai, H. Improve method for synthesis of 4-thioaryl-2, 3, 4-trihydro-1-benzo-thiopyrans: Acid-induced stereoselective intermolecular cycloaddition of α,β-unsaturated aldehydes with arenethiols. Synlett 2001, 1317–1319. [Google Scholar] [CrossRef]
- Jafarzadeh, M.; Amani, K.; Nikpour, F. Solvent-free and room temperature synthesis of thiochromans in presence of a catalytic amount of tungstophosphoric acid. Tetrahedron Lett. 2005, 46, 7567–7569. [Google Scholar] [CrossRef]
- Ishino, Y.; Nakamura, M.; Nishiguchi, I.; Hirashima, T. Facile synthesis of 3, 4-dihydro-2H-1-benzothiopyrans: Zinc iodide induced intermolecular cycloaddition between allyl alcohols and arenethiols. Synlett 1991, 633–635. [Google Scholar] [CrossRef]
- Chu, C.-M.; Gao, S.; Sastry, M.N.V.; Yao, C.-F. Iodine-catalysed Michael addition of mercaptans to α,β-unsaturated ketones under solvent-free conditions. Tetrahedron Lett. 2005, 46, 4971–4974. [Google Scholar] [CrossRef]
- Banerjee, A.K.; Vera, W.; Mora, H.; Laya, M.S.; Bedoya, L.; Cabrera, E.V. Iodine in organic synthesis. J. Sci. Ind. Res. 2006, 65, 299–308. [Google Scholar]
- Zhou, Y.; Yan, P.; Li, G.; Chen, Z. Progress in synthetic application of iodine as a Lewis acid catalyst. Chin. J. Org. Chem. 2009, 29, 1719–1727. [Google Scholar]
- Kabuto, K.; Kikuchi, Y.; Yamaguchi, S.; Inoue, N. The synthesis and stereochemistry of 4-chromanones and 4-chromanols with bulky substituents. Bull. Chem. Soc. Jpn. 1973, 46, 1839–1844. [Google Scholar] [CrossRef]
- Pouget, C.; Fagnere, C.; Basly, J.-P.; Leveque, H.; Chulia, A.-J. Synthesis and structure of flavan-4-ols and 4-methoxyflavans as new potential anticancer drugs. Tetrahedron 2000, 56, 6047–6052. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Button, M.A.C. Efficient syntheses of thiochromans via cationic cycloadditions. J. Org. Chem. 2001, 66, 5595–5600. [Google Scholar] [CrossRef] [PubMed]
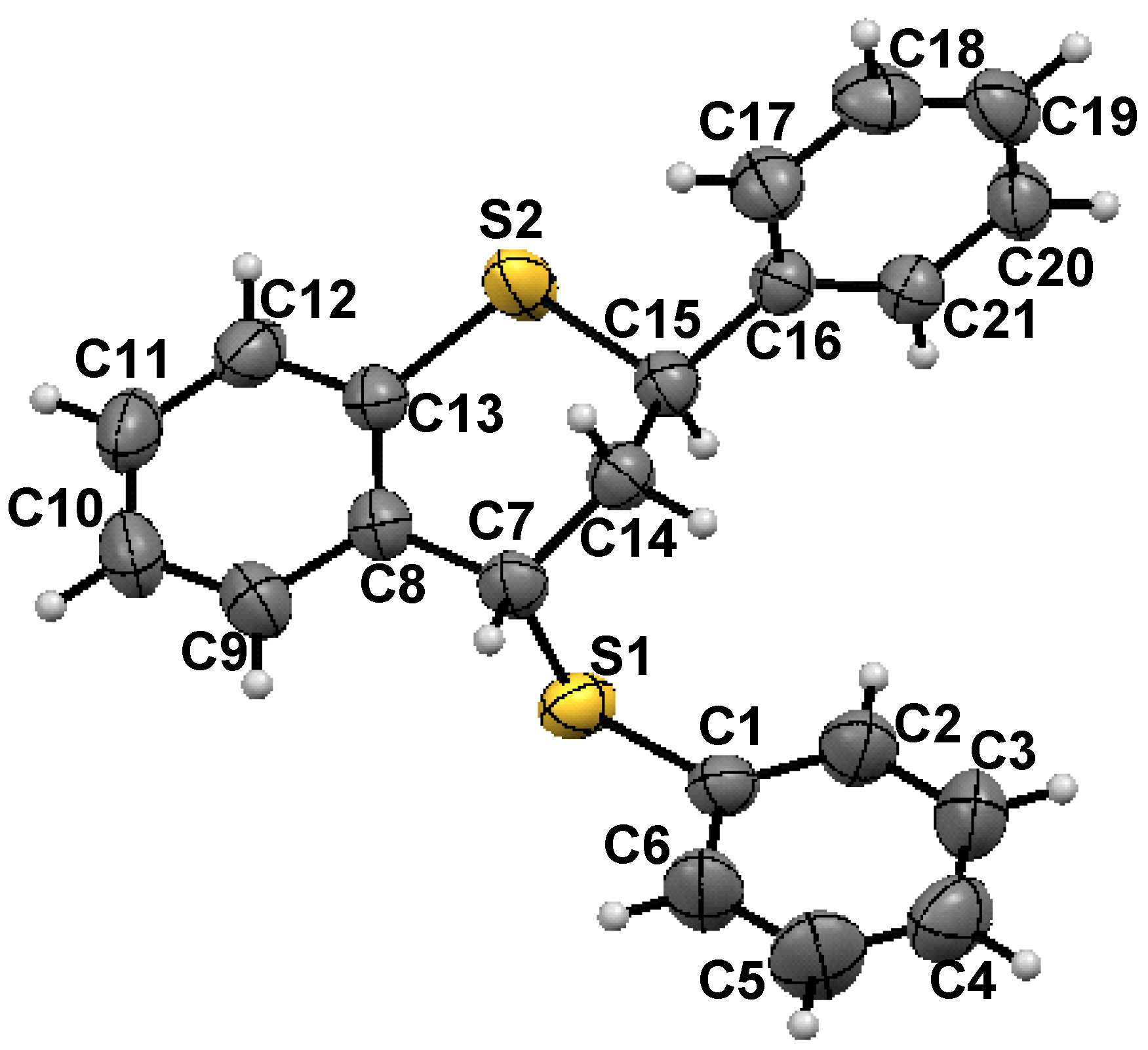
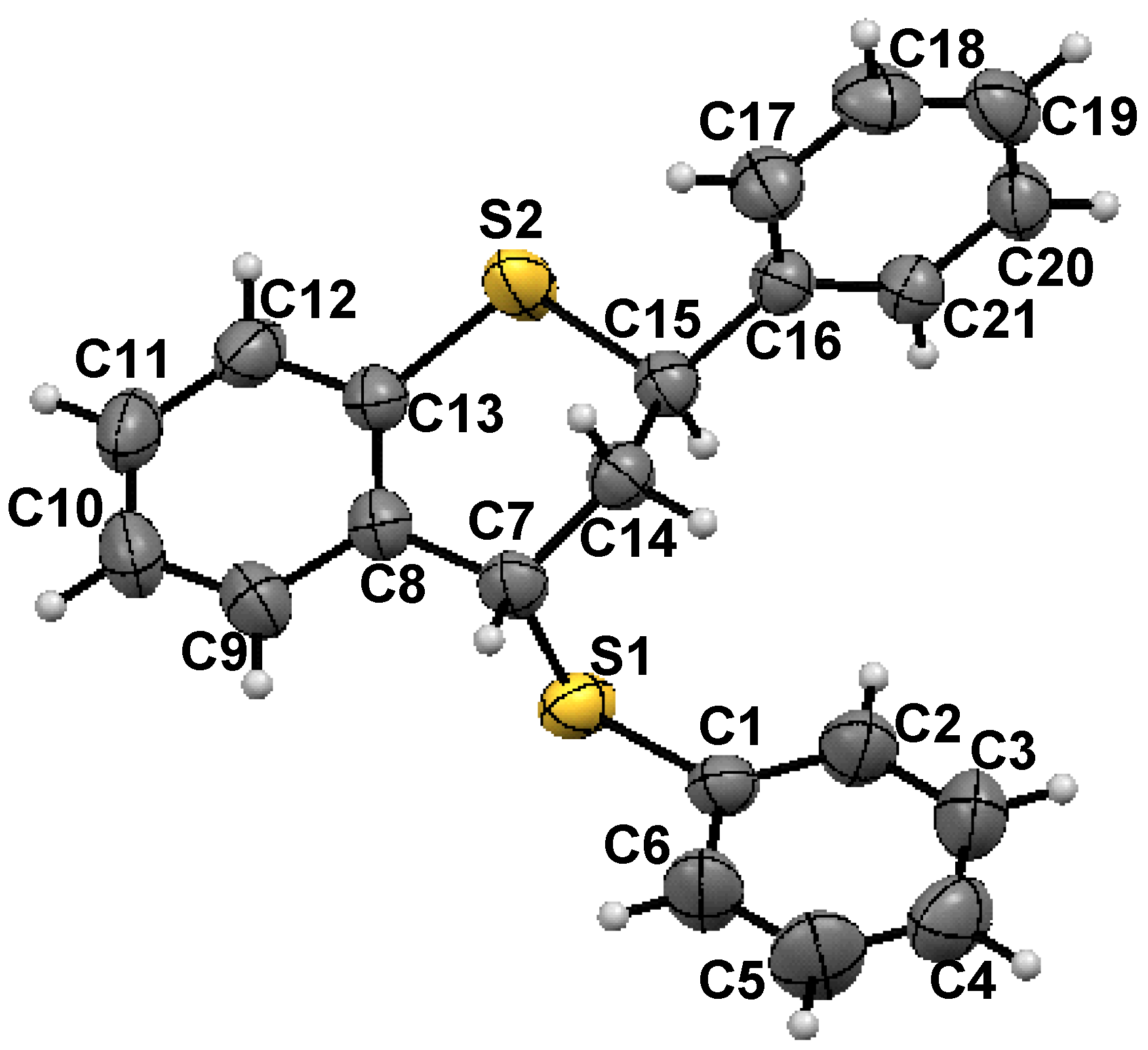
- X-ray crystallography: X-Ray single crystal data were collected using Mo Kα (λ = 0.7107 Å) radiation on a SMART APEX II diffractometer equipped with CCD area detector. Data collection, data reduction, structure solution/refinement were carried out using the software package of SMART APEX. The single crystal structure of 1 was solved by direct method and refined. The non hydrogen atoms were treated anisotropically. The positions of all hydrogen atoms were generated by their idealized geometry and refined using a riding model. Crystal dimension: 0.32 × 0.26 × 0.19 mm; T = 298(2) K; orthorhombic, space group Pccn ; a = 29.205(2), b = 9.3942(7), c = 12.8072(10) Ǻ; V = 3513.8(5) Ǻ3; Z = 8, ρcalcd = 1.265 gcm−3; µ = 0.300 mm−1; F(000) = 1408; θmin/max/° = 1.39/28.11; Rint = 0.0302; Range of h, k, l = -38/38, -11/12, -16/16; 36889 reflections collected of which 4268 were unique, 3273 observed [I > 2σ(I)] reflections, 208 parameters were refined; R1 = 0.0410, wR2 = 0.1032; Goodness of fit on F2 = 1.006; CCDC -799121 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc. cam.ac.uk /data request /cif.

© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mallik, A.K.; Mandal, T.K.; Pal, R.; Patra, A. trans-2-Phenyl-4-thiophenoxy-3,4-dihydro-2H-1-benzothiopyran. Molbank 2011, 2011, M719. https://doi.org/10.3390/M719
Mallik AK, Mandal TK, Pal R, Patra A. trans-2-Phenyl-4-thiophenoxy-3,4-dihydro-2H-1-benzothiopyran. Molbank. 2011; 2011(1):M719. https://doi.org/10.3390/M719
Chicago/Turabian StyleMallik, Asok K., Tapas K. Mandal, Rammohan Pal, and Amarendra Patra. 2011. "trans-2-Phenyl-4-thiophenoxy-3,4-dihydro-2H-1-benzothiopyran" Molbank 2011, no. 1: M719. https://doi.org/10.3390/M719
APA StyleMallik, A. K., Mandal, T. K., Pal, R., & Patra, A. (2011). trans-2-Phenyl-4-thiophenoxy-3,4-dihydro-2H-1-benzothiopyran. Molbank, 2011(1), M719. https://doi.org/10.3390/M719