Theoretical Characterization of Three 2,2-Diphenyl-1,3,2-oxazaborolidin-5-ones: Molecules with Fungicide Activities


Abstract
:1. Introduction
Computational Methodology:
2. Results and Analysis
3. Conclusions
Supplementary materials
Supplementary File 1Supplementary File 2Supplementary File 3Supplementary File 4Supplementary File 5Supplementary File 6Supplementary File 7Supplementary File 8Supplementary File 9Acknowledgements
References and Notes
- Velasco, B.; Trujillo-Ferrara, J.G.; Fabila-Castillo, L.H.; Miranda, R.; Sanchez-Torres, L.E. In vitro apoptotic activity of 2,2-diphenyl-1,3,2-oxazaborolidin-5-ones in L5178Y cells. Life Sci. 2007, 80, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Anaissie, E.J. Clinical Mycology; Churchill Livingstone: Philadelphia, 2003; pp. 195–239. [Google Scholar]
- Groziak, M.P. Boron therapeutics on the horizons. Am. J. Therapeutics. 2001, 8, 321–328. [Google Scholar] [CrossRef]
- Dobrydneva, Y.; Abelt, C.J.; Dovel, B.; Thadigiri, C.M.; Williams, R.L.; Blackmore, P.F. 2-Aminoethoxydiphenyl borate as a prototype drug for a group of structurally related calcium channel blockers in human platelets. Mol. Pharmacol. 2006, 69, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Flückiger, R.; Henson, E.; Hess, G.; Gallop, P. Mass spectral and HPLC analysis of biological compounds with diphenylborinic acid. Biomed. Mass Specrometry. 1984, 11, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Strang, C.J.; Henson, E.; Okamoto, Y.; Paz, M.A.; Gallop, P.M. Separation and determination of α-amino acids by boroxazolidones formation. Analit. Biochem. 1989, 178, 276–286. [Google Scholar] [CrossRef]
- Velasco-Bejarano, B.; Trujillo-Ferrara, J.G.; Miranda, R. Preparation of apoptotic inducers 2,2-diphenyl-1,3,2-oxazaborolidin-5-ones under alkaline conditions. SYNLETT 2007, 6, 921–924. [Google Scholar] [CrossRef]
- Nicolás, I.; Vilchis, M. B.; Aragón, N.; Miranda, R.; Hojer, G. A.; Castro, M. Theoretical study of the structure and antimicrobial activity of horminone. Int. J. Quantum Chem. 2003, 93, 411–421. [Google Scholar] [CrossRef]
- Nicolás, I.; Castro, M. Theoretical study of the complexes of horminone with Mg2+ and Ca2+ ions and their relation with the bacteriostatic activity. J. Phys. Chem. A 2006, 110, 4564–4573. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Albores, A.; Nicolás-Vázquez, I.; Miranda-Ruvalcaba, R.; Moreno-Martínez, E. Mass spectrometry/mass spectrometry study on the degradation of B-aflatoxins in maize with aqueous citric acid lactonic moiety. Am. J. Agril. & Biol. Sci. 2008, 3, 482–489. [Google Scholar]
- Deppmeier, B.J.; et al. Spartan '02, Wavefunction Inc.: Irvine CA, 2001.
- Hehre, W. J. A Guide to Molecular Mechanics & Quantum Chemical Calculations. Wavefunction, Inc.: Irvine, CA, USA, 2003; pp. 61–88. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Phys. Chem. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Frisch, M. J.; et al. Gaussian 03, Revision D.01; Gaussian, Inc.: Wallingford CT, 2004.
- Farfán, N.; Joseph-Nathan, P.; Chiquete, L.M.; Contreras, R. Syntheses and structures of two new dibenzobicyclic phenylboronates. J. Organomet. Chem. 1988, 348, 149–156. [Google Scholar] [CrossRef]
- Farfán, N.; Mancilla, T.; Castillo, D.; Uribe, G.; Carrillo, L.; Joseph-Nathan, P.; Contreras, R. NMR and X-ray diffraction studies of two bicyclic borates containing chiral boron and nitrogen atoms. J. Organomet. Chem. 1990, 381, 1–13. [Google Scholar] [CrossRef]
- Farfán, N.; Castillo, D.; Joseph-Nathan, P.; Contreras, R.; Szentpály, L.V. Through-bond modulation of N→B ring formation shown by NMR and X-ray diffraction studies of borate derivatives of pyridyl alcohols. J. Chem. Soc., Perkin Trans. 1992, 2, 527–532. [Google Scholar] [CrossRef]
- Toyota, S.; Oki, M. Structure of intramolecular boron-amine complexes and proposal of tetrahedral character for correlation between molecular structure and barrier to the N-B bonds. Bull Chem. Soc. Jpn. 1992, 65, 1832–1840. [Google Scholar] [CrossRef]
- Kliegel, W.; Lubkowitz, G.; Rettig, S.J.; Trotter, J. Structural studies of organoboron compounds. XLIX14,6-Bis(l-phenyl-2-nitroethyl)-2-(4-methoxyphenyl)-l,3-dioxa-4,6-diaza-2-boracyclohexane. Can. J. Chem. 1991, 69, 1227–1232. [Google Scholar] [CrossRef]
- HÖpfl, H.; Farfán, N.; Castillo, D.; Santillan, R.; Contreras, R.; Martínez-Martínez, F. J.; Galván, M.; Alvarez, R.; Fernández, L.; Halut, S.; et al. Dynamic NMR and X-ray diffraction study of (N-B) -diphenyl(2-aminoethoxy) borane derivatives of ephedrines, and pseudoephedrines. J. Organomet. Chem. 1997, 544, 175–188. [Google Scholar] [CrossRef]
- HÖpfl, H.; Pérez, N.; Rojas, S.; Santillan, R.; Farfán, N. X-ray crystallographic study of neutral boron chelates with b-diketones and tropolone. Heteroat. Chem. 1998, 9, 359–368. [Google Scholar] [CrossRef]
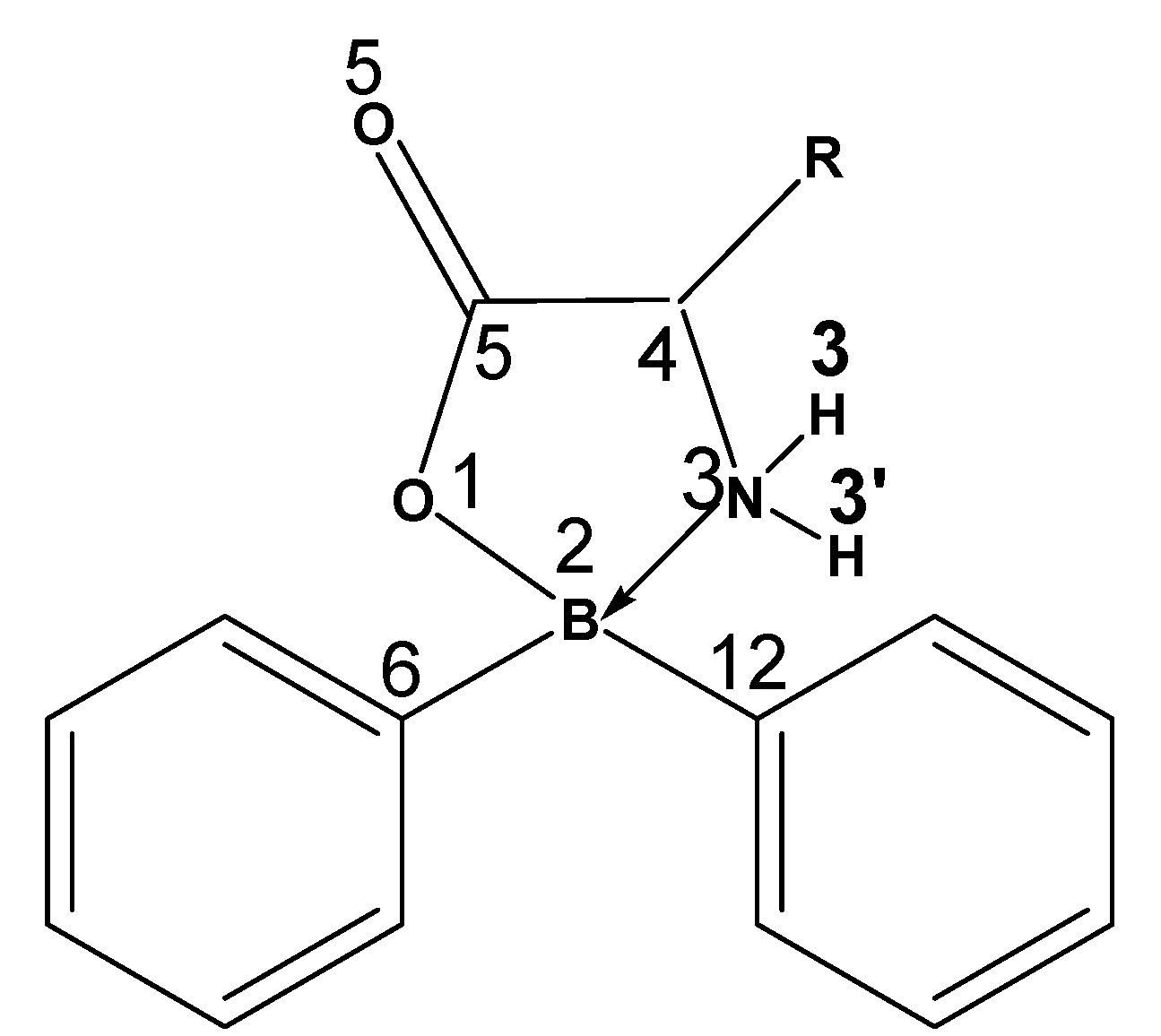
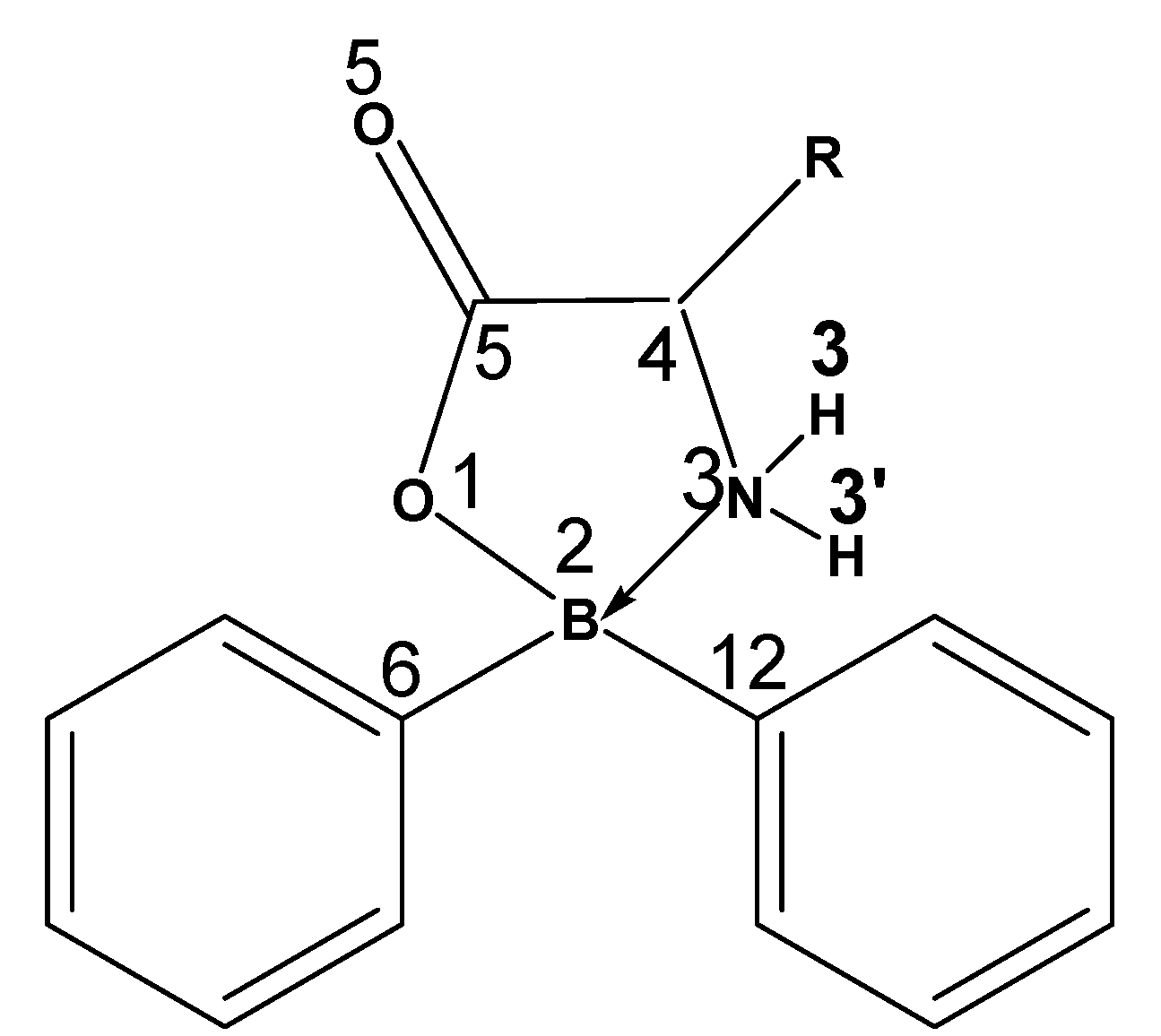
| Compound | R= α-amino acid residue |
| 1 | Gly: H |
2 | His: 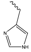 |
| 3 | Thr: 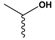 |

{kind=link}
| Compound* | Aspergillus niger 16409 | Cryptococcus neoformans | C.albicans 5609 | C. parapsilosis 2019 | C. krusei 6258 |
| 1 | +++ | ++++ | +++ | ++++ | ++++ |
| 2 | ++ | +++ | ++ | ++ | ++ |
| 3 | ++ | +++ | +++ | +++ | +++ |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Vilchis, M.; Velasco, B.; Penieres, G.; Cruz, T.; Miranda, R.; Nicolás, I. Theoretical Characterization of Three 2,2-Diphenyl-1,3,2-oxazaborolidin-5-ones: Molecules with Fungicide Activities. Molbank 2009, 2009, M600. https://doi.org/10.3390/M600
Vilchis M, Velasco B, Penieres G, Cruz T, Miranda R, Nicolás I. Theoretical Characterization of Three 2,2-Diphenyl-1,3,2-oxazaborolidin-5-ones: Molecules with Fungicide Activities. Molbank. 2009; 2009(2):M600. https://doi.org/10.3390/M600
Chicago/Turabian StyleVilchis, Martha, Benjamin Velasco, Guillermo Penieres, Tonatiuh Cruz, René Miranda, and Inés Nicolás. 2009. "Theoretical Characterization of Three 2,2-Diphenyl-1,3,2-oxazaborolidin-5-ones: Molecules with Fungicide Activities" Molbank 2009, no. 2: M600. https://doi.org/10.3390/M600
APA StyleVilchis, M., Velasco, B., Penieres, G., Cruz, T., Miranda, R., & Nicolás, I. (2009). Theoretical Characterization of Three 2,2-Diphenyl-1,3,2-oxazaborolidin-5-ones: Molecules with Fungicide Activities. Molbank, 2009(2), M600. https://doi.org/10.3390/M600