Experimental
Melting points were determined on a hot stage instrument and are uncorrected. Infrared spectra were recorded as KBr pellets on a Perkin Elmer System 2000 FTIR. 1H-NMR spectra were recorded on a Bruker AMX300 spectrometer at 300 MHz and chemical shifts are expressed in ppm using TMS as internal standard. 13C-NMR spectra were recorded on a Bruker AMX300 spectrometer at 75.4 MHz and chemical shifts are expressed in ppm using residual solvent signal as internal standard. Assignment of carbon absorptions were confirmed by DEPT experiments in all cases. Mass spectra were recorded on a Varian CP-3800 GC system with a Saturn 2200 MS station.
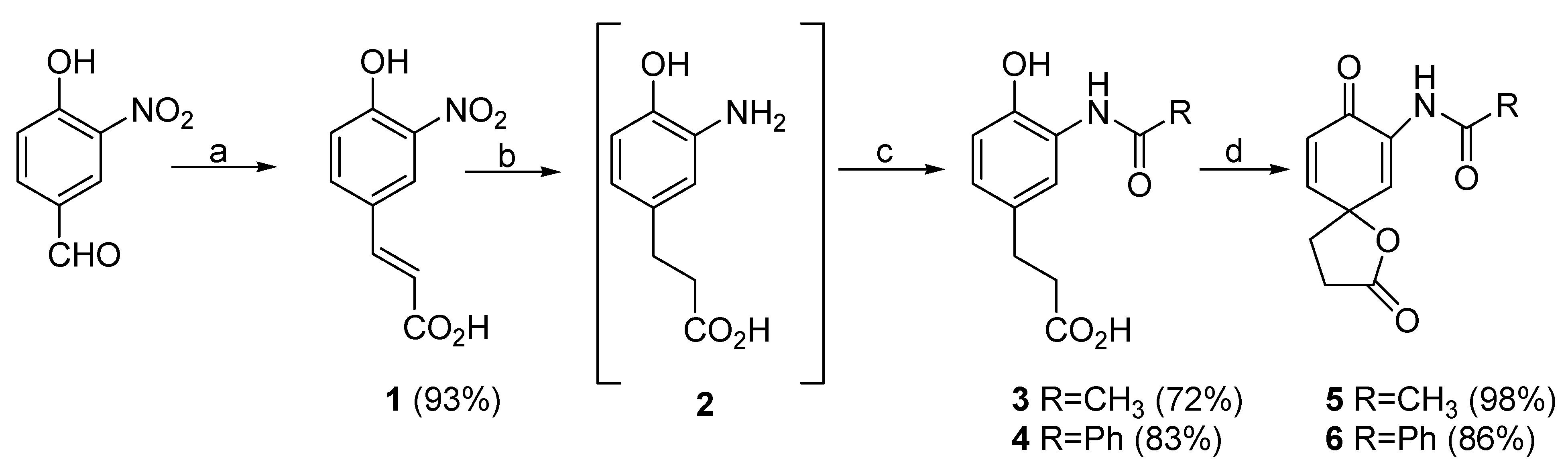
(2E)-3-(4-Hydroxy-3-nitrophenyl)acrylic acid (1):
To a solution of 4-hydroxy-3-nitrobenzaldehyde (1.073 g, 6.43 mmol) in pyridine (25 mL) was added piperidine (25 drops) and malonic acid (1.671 g, 16.1 mmol). The resulting mixture was stirred at 65
oC overnight. The reaction was cooled and acidified to pH=2 with 50% HCl, resulting in the formation of a yellow solid. This mixture was extracted with ethyl acetate (2 x 150 mL), the organic fractions were combined, washed with brine (150 mL), dried (MgSO
4), and the solvent was evaporated to afford a yellow solid (1.250 g, 93%). No further purification was carried out on this material. mp: 227-228
oC. IR (KBr) cm
-1: 2942 (OH), 1684 (CO
2H), 1626 (C=C), 1533,1270 (NO
2).
1H-NMR (acetone-d
6) δ: 2.87 (broad s, 1H, OH), 6.58 (d, 1H,
J=16.0 Hz, H-2), 7.27 (d, 1H,
J=8.8 Hz, H-8), 7.70 (d, 1H,
J=16.0 Hz, H-3), 8.08 (dd, 1H,
J=2.2, 8.8 Hz, H-9), 8.40 (d, 1H,
J=2.2 Hz, H-5), 10.67 (broad s, 1H, CO
2H). The
1H-NMR spectrum of this compound is in agreement with the previously published data [
4].
3-(3-Acetamido-4-hydroxyphenyl)propanoic acid (3):
To a solution of 1 (210 mg, 1.00 mmol) in tetrahydrofuran (20 mL) was added 10% Pd/C (32 mg). The resulting mixture was placed in a hydrogenator, flushed (5 times) with hydrogen, and left to agitate under pressure (39 psi) for 6 hrs. The reaction mixture was filtered through Celite ® (rinsing with tetrahydrofuran) and the solvent was evaporated to give 2 as an off-white solid. The structure of 2 was established only on the basis of its 1H-NMR data, and the crude reaction product was used without further purification. 1H-NMR (methanol-d4) δ: 2.46 (t, 2H, J=8.0 Hz , H-3), 2.69 (t, 2H, J=8.0 Hz, H-2), 6.48 (m, 1H, H-5), 6.60 (m, 2H, H-8, H-9).
To a solution of 2 in tetrahydrofuran (20 mL) was added acetyl chloride (79 mg, 1.13 mmol) and the solution was stired at room temperature for 60 minutes. Water (15 mL) was added and the mixture was extracted with ethyl acetate (2 x 50 mL). The organic fractions were combined, washed with brine (50 mL), dried (MgSO4), and the solvent was evaporated to afford an off-white solid. The product was recrystallized from hexane/acetone to afford a white solid (104 mg) in 72% yield from 1. mp: 169-170oC. IR (KBr) cm-1: 3393 (NH, OH), 1699 (CO2H), 1657 (NHAc). 1H-NMR (acetonitrile-d3) δ: 2.15 (s, 3H, H-2'), 2.54 (t, 2H, J=7.5 Hz, H-3), 2.78 (t, 2H, J=7.5 Hz, H-2), 6.82 (d, 1H, J=8.3 Hz, H-8), 6.95 (dd, 1H, J=2.1, 8.3 Hz, H-9), 7.06 (d, 1H, J=2.1 Hz, H-5), 8.53 (broad s, 1H, OH), 8.81 (s, 1H, NH). 13C-NMR (acetonitrile-d3) δ: 23.5 (C-2'), 31.0 (C-3), 36.0 (C-2), 119.5 (C-5), 123.0 (C-8), 127.1 (C-6), 127.3 (C-9), 133.7 (C-4), 147.9 (C-7), 172.1 (C-1), 174.4 (C-1'). MS m/e (rel. %): 223 [M+] (> 1%), 206 (23), 205 (36), 159 (17), 146 (100), 105 (11).
3-(3-Benzamido-4-hydroxyphenyl)propanoic acid (4):
To a solution of 1 (222 mg, 1.06 mmol) in tetrahydrofuran (20 mL) was added 10% Pd/C (33 mg). The resulting mixture was then placed in a hydrogenator, flushed (5 times) with hydrogen and left to agitate under pressure (36 psi.) overnight. The reaction mixture was filtered through Celite ® (rinsing with tetrahydrofuran) and the solvent was evaporated to afford 2 which was used without further purification.
To a solution of 2 in tetrahydrofuran (20 mL) was added benzoyl chloride (154 mg, 1.1 mmol) and the solution was stirred at room temperature for 30 minutes. 10% HCl (25 mL) was added, the mixture was stirred for 5 minutes and extracted with dichloromethane (2 x 35 mL). The organic fractions were combined, dried (MgSO4), and the solvents were evaporated to afford an off-white solid. The crude product was recrystallized with hexane/acetone to afford an off-white solid (250 mg) in 83% yield from 1. mp: 206-207oC. IR (KBr) cm-1: 3201 (NH, OH), 1692 (CO2H), 1636 (NHBz). 1H-NMR (acetone-d6) δ: 2.60 (t, 2H, J=7.9 Hz, H-3), 2.84 (t, 2H, J=7.9 Hz, H-2), 6.89 (d, 1H, J=8.2 Hz, H-8), 7.00 (dd, 1H, J=2.1, 8.2 Hz, H-9), 7.57 (m, 4H, H-5, H-4', H-5', H-6'), 8.05 (d, 2H, J=8.2 Hz, H-3', H-7'), 9.07 (broad s, 1H, NH), 9.54 (broad s, 1H, OH), 10.58 (broad s, 1H, CO2H). 13C-NMR (acetone-d6) δ: 30.9 (C-3), 36.2 (C-2), 118.7 (C-8), 123.3 (C-5), 123.4 (C-6), 126.9 (C-9), 127.4 (C-4), 128.5 (C-4', C-6'), 129.6 (C-3', C-7'), 133.0 (C-5'), 135.0 (C-2'), 148.0 (C-7), 167.3 (C-1'), 173.9 (C-1). MS m/e (rel. %): 285 [M+] (12), 267 (100), 221 (16), 208 (95), 146 (24), 77 (25).
N-(2,8-Dioxo-1-oxaspiro[4.5]deca-6,9-dien-7-yl)acetamide (5):
To a cold (0oC) solution of 3 (122 mg, 0.55 mmol) in acetone (10 mL) was added phenyliodine (III) bistrifluoroacetate (PIFA) (306 mg, 0.71 mmol) in one portion and the resulting solution was stirred at 0oC for 25 minutes. The reaction mixture was diluted with ethyl acetate (20 mL), and washed with cold water (10 mL). The organic fraction was separated, dried (MgSO4) and evaporated to afford a beige solid. The crude product was triturated from chloroform affording an off-white solid (120 mg) in 98% yield. mp: 116oC (decomposes). IR (KBr) cm-1: 3333 (NH), 1777 (lactone), 1668 (amide), 1650 (ketone). 1H-NMR (chloroform-d) δ: 2.17 (s, 3H, H-2'), 2.44 (m, 2H, H-4), 2.81 (m, 2H, H-3), 6.35 (d, 1H, J=10.0 Hz, H-9), 6.94 (dd, 1H, J=3.1, 10.0 Hz, H-10), 7.75 (d, 1H, J=3.1 Hz, H-6), 7.99 (broad s, 1H, NH). 13C-NMR (chloroform-d) δ: 24.9 (C-2'), 28.4 (C-4), 32.9 (C-3), 79.8 (C-5), 124.3 (C-6), 127.1 (C-9), 131.6 (C-7), 148.4 (C-10), 169.5 (C-1'), 175.5 (C-2), 179.4 (C-8). MS m/e (rel.%): 221 [M+] (> 1%), 206 (5), 178 (12), 177 (32), 143 (5), 136 (8), 135 (100), 106 (21).
N-(2,8-Dioxo-1-oxaspiro[4.5]deca-6,9-dien-7-yl)benzamide (6):
To a cold (0oC) solution of 4 (262 mg, 0.92 mmol) in acetone (30 mL) was added PIFA (396 mg, 0.92 mmol) in one portion and the solution was stirred at 0oC for 25 minutes. The reaction mixture was diluted with ethyl acetate (50 mL), and washed with cold water (20 mL). The organic fraction was separated, dried (MgSO4) and evaporated to afford a beige solid. Purification by column chromatography using 40% ethyl acetate/hexane as eluent afforded an off-white solid (243 mg) in 86% yield. mp: 137-138oC. IR (KBr) cm-1: 3381 (NH), 1781 (lactone), 1665 (amide), 1650 (ketone) 1H-NMR (chloroform-d) δ: 2.49 (m, 2H, H-4), 2.84 (m, 2H, H-3), 6.42 (d, 1H, J=10.0 Hz, H-9), 6.99 (dd, 1H, J=3.1, 10.0 Hz, H-10), 7.54 (m, 3H, H-4', H-5', H-6'), 7.86 (m, 2H, H-3', H-7'), 7.95 (d, 1H, J=3.1 Hz, H-6), 8.81 (broad s, 1H, NH). 13C-NMR (chloroform-d) δ: 28.4 (C-4), 33.0 (C-3), 79.8 (C-5), 124.5 (C-6), 127.2 (C-9), 127.3 (C-4', C-6'), 129.2 (C-3', C-7'), 131.7 (C-7), 132.7 (C-5'), 133.9 (C-2'), 148.6 (C-10), 166.2 (C-1'), 175.4 (C-2), 179.7 (C-8). MS m/e (rel.%): 283 [M+] (> 1%), 239 (25), 106 (11), 105 (100), 77 (49), 51 (14).

{kind=link}